
吉西他滨类似物的设计、合成及体外抗肿瘤活性
2020-05-13 08:26徐广凌张小怡王晨阳端木彦涛王幸幸李茹君赵伟栋
合成化学 2020年4期
徐广凌, 张小怡, 王晨阳, 端木彦涛, 王幸幸, 李茹君, 从 梅*, 赵伟栋*
(1. 新乡医学院 a. 医学检验学院; b. 药学院,河南 新乡 453003)
癌症是威胁人类健康的重大疾病,已成为全世界最严重的公共卫生问题之一[1]。化疗在癌症的治疗中起着非常重要的作用,是临床治疗癌症不可或缺的方案之一。然而,由于化疗药物自身分子结构的局限,在临床应用中面临众多挑战[2]。如吉西他滨(Gem, Chart 1),1996年经FDA批准进入市场,通过干扰DNA的合成发挥抑制肿瘤生长的作用,是治疗晚期非小细胞肺癌、胰腺癌的一线药物,也可用于乳腺癌、卵巢癌、鼻咽癌等恶性肿瘤的治疗[3]。然而,吉西他滨极易被肝脏和血液中的脱氧胞苷脱氨酶(CDA)脱去4-位氨基生成无活性产物2,2′-双氟脱氧尿嘧啶核苷(dFdU, Chart 1),导致其在人体内半衰期极短(<17 min)[3]。因此,临床通常需持续的静脉给药或联合用药来维持其细胞毒性作用。然而,这又增加了药物对身体正常组织器官的毒副作用,临床表现为骨髓抑制、肾毒性、胃肠道反应等[3]。此外,吉西他滨为亲水性药物,难以通过自由扩散透过细胞膜,必须借助相应的核苷转运体(如hENT)将其转入细胞[3]。但这种转运体在多数病人(>65%)体内表现为缺失或表达量低[4],进一步限制了吉西他滨的药效。此外,转运体问题也是肿瘤细胞对吉西他滨产生耐药的主要原因之一[5]。
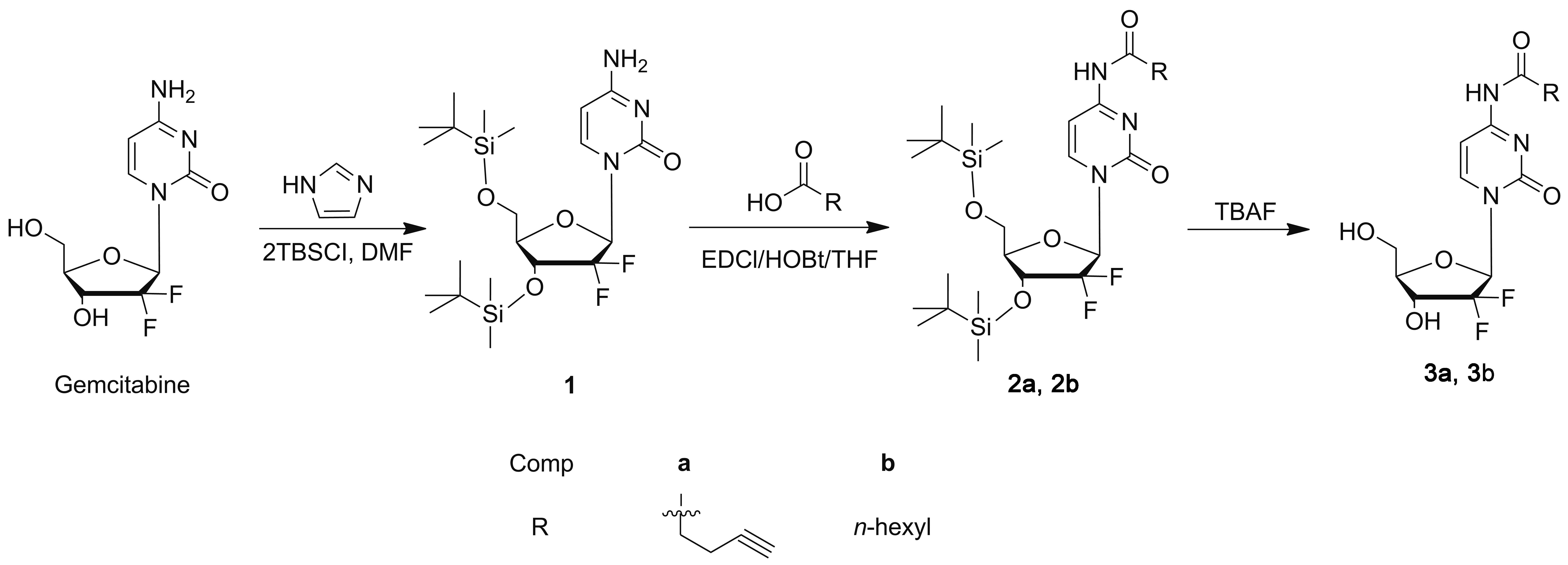
Scheme 1
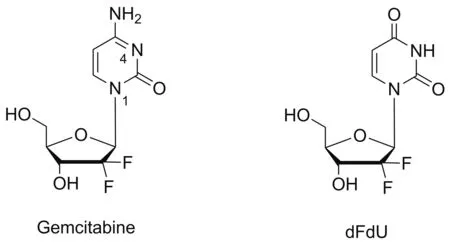
Chart 1
为此,研究人员对吉西他滨的结构进行了修饰和改造,取得了一定进展[6-7]。近年来,纳米药物递送系统为克服吉西他滨类化疗药物存在的弊端提供了策略[8-9]。利用纳米技术构建的小分子化疗药物递送系统,通过增加生物膜的通透性,控制药物释放,延长血液循环时间,提高负载药物生物利用度[10-11];另外,肿瘤组织中血管丰富、血管壁间隙相对较宽、结构完整性差、淋巴回流缺失,纳米颗粒可利用肿瘤增强的渗透和滞留效应(EPR)有效地蓄积于肿瘤区域,从而增强药物在体内分布的特异性,提高化疗效果及降低毒副作用[9]。Doxil是一种PEG化阿霉素脂质体,是最早批准的用于临床治疗的纳米药物[12]。Doxil可延长阿霉素的体内循环时间,并增加其在肿瘤部位的浓度,从而降低在正常组织中的浓度。例如,Doxil可以显著地降低阿霉素的心脏毒性,并在多种恶性肿瘤疾病的治疗中表现出与阿霉素类似的抑瘤效果[13]。Abraxan是一种白蛋白和紫杉醇结合形成的纳米颗粒,既能保持紫杉醇的肿瘤治疗效果又能避免传统紫杉醇制剂Taxol因助溶剂Cremophor El而引起的毒副作用。研究表明[14],给予相同紫杉醇剂量的Abraxane和Taxol, Abraxane在肿瘤部位的浓度较Taxol更高,即Abraxane能通过EPR效应更多的在肿瘤部位富集。
将药物分子被动地包裹在载体中,除少数可能产生的疏水相互作用或静电作用,药物分子和载体之间通常没有较强的作用力使之连接。因此,大部分药物载体存在药物包裹率低、易渗漏的问题,在制剂贮存阶段以及体内循环时都有可能发生,特别是对于吉西他滨类极性药物,很难直接将其包载在载体材料中构建纳米递送系统。
基于上述研究,本文将戊炔酸或正庚酸和吉西他滨通过酰胺键偶联获得目标化合物2a, 2b和3a, 3b(Scheme 1),期望能发现一种制备亲脂性吉西他滨衍生物的路线,并获得高活性抗肿瘤的候选化合物,为临床借助纳米技术构建高稳定性、低毒性的药物递送系统奠定基础。
1 实验部分
1.1 仪器与试剂
SGWRX-4型显微熔点仪;Bruker Ascend-400 MHz型核磁共振仪(CDCl3或CD3OD-d4为溶剂,TMS为内标);Bruker 7-Tesla FT-ICR型质谱仪。
吉西他滨,>99%,阿法埃莎公司;1-羟基苯并三唑(HOBt),99%,伊诺凯公司;1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCl), 99%,麦克林公司;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 5′-OH保护的Gem类似物(1)的合成
在干燥的圆底烧瓶中加入Gem 263.0 mg(1.0 mmol)和DMF 4.0 mL,搅拌使固体溶解;依次加入叔丁基二甲基氯硅烷602.4 mg(4.0 mmol)和咪唑273.9 mg(4.0 mmol),反应至终点(TLC监测)。将反应液缓慢倾至蒸馏水(30 mL)中,用乙酸乙酯(3×15 mL)萃取,合并有机相,经无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析(洗脱剂:A=二氯甲烷/甲醇=100/3,V/V)纯化得白色粉末1,收率63.3%, m.p.118~120 ℃;1H NMR(400 MHz, CDCl3)δ: 7.68(d,J=7.6 Hz, 1H), 6.34~6.31(m, 2H), 5.70(d,J=7.6 Hz, 1H), 4.34~4.26(m, 1H), 4.00~3.97(m, 1H), 3.90~3.88(m, 1H), 3.81~3.77(m,1H), 0.94~0.90(m, 18H), 0.11(m, 12H);13C NMR(100 MHz, CD3OD)δ: 165.8, 155.6, 140.6, 122.0(t,J=258.3 Hz, CF2), 95.1, 84.5~83.9(m), 80.9~80.8(m), 70.0~69.6(m), 60.1, 29.7, 25.8, 25.5, 18.3, 18.0, -4.75, -5.29, -5.46, -5.45; HR-MS(ESI)m/z: Calcd for C21H39N3O4F2Si2{[M+H]+}492.2525, found 492.2521。
(2) 2a, 2b的合成(以2a为例)
在冰浴条件下,依次向圆底烧瓶中加入THF 2.5 mL, 4-戊酸59.2 mg(0.6 mmol), EDCl 192.0 mg(1.0 mmol)和HOBt 135.4 mg(1.0 mmol),反应30 min;加入中间产物1 244.2 mg(0.5 mmol),搅拌下继续反应24 h。旋蒸除溶,残余物用水(15 mL)溶解,用乙酸乙酯(3×15 mL)萃取,合并有机相,经无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析(洗脱剂:B=乙酸乙酯/石油醚=1/2,V/V)纯化得化合物2a。
以正庚酸代替戊炔酸,用类似的方法合成2b。
2a: 白色固体,收率33.0%, m.p.97~99 ℃;1H NMR(400 MHz, CDCl3)δ: 9.43(br s, 1H), 7.60(d,J=7.6 Hz, 1H), 7.43(d,J=8.0 Hz, 1H), 6.35~6.32(m, 1H), 4.36~4.30(m, 1H), 4.05~3.95(m, 2H), 3.83~3.80(m, 1H), 2.75(t,J=7.0 Hz, 2H), 2.59~2.55(m, 2H), 2.02(t,J=2.8 Hz, 1H), 0.96(s, 9H), 0.91(s, 9H), 0.14~0.11(m, 12H);13C NMR(100 MHz, CDCl3)δ: 172.1, 163.1, 154.8, 144.1, 122.9(t,JC-F=259.5 Hz, CF2), 97.2, 85.1~84.4(m), 82.3, 81.5~81.4(m), 69.6~69.2(m), 59.9, 53.4, 36.1, 25.9, 25.5, 18.3, 18.0, 14.0, -4.8, -5.3, -5.4; HR-MS(ESI)m/z: Calcd for C26H43N3O5F2Si2{[M+Na]+}594.2607, found 594.2603。
2b: 白色固体,收率51.0%, m.p.49~51 ℃;1H NMR(400 MHz, CDCl3)δ: 8.08(d,J=7.6 Hz, 1H), 7.49(d,J=7.6 Hz, 1H), 6.34~6.30(m, 1H), 4.37~4.29(m, 1H), 4.03~3.95(m, 1H), 3.83~3.79(m, 2H), 2.48(t,J=7.4 Hz, 2H), 2.36(t,J=7.4 Hz, 2H), 1.68~1.63(m, 4H), 0.95(s, 9H), 0.90~0.87(m, 15H), 0.13~0.10(m, 12H);13C NMR(100 MHz, CDCl3)δ: 179.4, 174.2, 163.0, 144.5, 123.1 (t,JCF=258.9 Hz, CF2), 96.7, 81.5~81.4(m), 69.7~69.2(m), 59.9, 37.6, 34.2, 31.4, 28.8, 28.7, 25.8,25.4, 24,8, 2.7, 22.5, 22.4, 18.3, 17.9, 14.0, -4.8, -5.3, -5.5; HR-MS(ESI)m/z: Calcd for C28H52N3O5F2Si2{[M+H]+}604.3414, found 604.3411。
(3) 3a, 3b的合成通法
在干燥的圆底烧瓶中加入中间产物2a或2b 113 mg(0.20 mmol), TBAF 0.50 mL(0.50 mmol)和超干THF 3.0 mL,搅拌下反应20 min。浓缩,残余物经硅胶柱层析纯化得产物3a或3b。
3a: 白色固体,收率58.1%, m.p.147~149 ℃;1H NMR(400 MHz, CD3OD)δ: 8.35(d,J=7.6 Hz, 1H), 7.49(d,J=7.6 Hz, 1H), 6.26(t,J=7.2 Hz, 1H), 4.34~4.26(m, 1H), 3.99~3.94(m, 2H), 3.83~3.79(m, 1H), 2.69(t,J=7.2 Hz, 2H), 2.54~2.50(m, 2H), 2.28(t,J=2.6 Hz, 1H);13C NMR (100 MHz, CD3OD)δ: 173.8, 164.8, 157.7, 146.0, 124.0(t,J=257.5 Hz, CF2), 98.3, 86.9~86.2(m), 83.3, 82.9~82.8(m), 70.4, 70.2~69.9(m), 60.3, 37.1, 14.7; HR-MS(ESI)m/z: Calcd for C14H15N3O5F2{[M+Na]+}366.0877, found 366.0872。
3b: 白色固体,收率55%, m.p.116~118 ℃;1H NMR(400 MHz, CD3OD)δ: 8.33(d,J=7.6 Hz, 1H), 7.49(d,J=7.6 Hz, 1H), 6.26(t,J=7.2 Hz, 1H), 4.34~4.26(m, 1H), 3.99~3.94(m, 2H), 3.83~3.79(m, 1H), 3.31~3.30(m, 1H), 2.45 (t,J=7.6 Hz, 2H), 1.68~1.62(m, 2H), 1.42~1.28(m, 6H), 0.91(t,J=7.0 Hz, 3H);13C NMR(100 MHz, CD3OD)δ: 176.0, 164.9, 157.7, 146.0, 125.2(t,J=257.2 Hz, CF2), 98.3, 86.8, 86.2, 82.9~82.8(m), 70.4, 70.2~69.7(m), 60.3, 38.2, 32.7, 29.9, 26.0, 23.7, 14.4; HR-MS(ESI)m/z: Calcd for C16H23N3O5F2Si2{[M+Na]+}:398.1503, found 398.1499。
1.3 生物活性测试
(1) 体外抗肿瘤活性
将肿瘤细胞制成单细胞悬液,注入96孔细胞培养板中,每孔约6000个细胞。置入细胞培养箱中,于37 ℃, 5%CO2条件下过夜培养。先用DMSO将测试化合物溶解到合适浓度,再用DMEM培养基稀释到预设浓度。将96孔细胞培养板中的细胞培养基置换成含50 μM有效吉西他滨药物浓度的培养基,相同条件下继续培养72 h。然后将96孔细胞培养板中的含药培养基置换成100 μL的CCK-8稀释液(增强型CCK-8试剂盒,Beyotime Biotechnology, #C0042), 37 ℃于细胞培养箱中孵育1 h,即刻用酶标仪于450 nm波长处读取吸光度A值。设置相应量细胞培养液和CCK-8溶液但没有加入细胞的孔作为空白对照,计算各实验组的细胞活力。用DMEM细胞培养基将合成的化合物作系列浓度稀释(50 μM, 5 μM, 2.5 μM, 0.5 μM, 0.05 μM),以上述CCK-8试剂盒测定药物各浓度对肿瘤细胞的抑制活性,使用GraphPad Prism 7.0软件,采用非线性曲线拟合方法计算药物对肿瘤细胞的半数生长抑制率IC50值。
(2) 酶稳定性
用微量移液枪分别取150 μL含吉西他滨(100 μM)溶液或吉西他滨类似物3a(100 μM)的Tris-HCL缓冲溶液(pH 7.5, 50 mM),加入96孔细胞培养板中,然后在每个孔中加入50 μL浓度为0.005 μg/μLCDA酶(Abcam #99441),混匀后振荡2 min。使用酶标仪在280 nm波长下,测定每孔在反应开始后第1 min, 2 min, 3 min, 4 min, 5 min, 20 min, 45 min时的吸光度值[15]。
2 结果与讨论
2.1 体外肿瘤细胞增殖抑制活性
采用CCK-8法对化合物2a, 2b, 3a和3b的体外抗肿瘤活性进行了测试。结果表明,在高测试浓度下(50 μM),化合物2a和2b对人胰腺癌细胞MiaPaCa2, SW1990, PANC-1和人乳腺癌细胞MCF-7细胞均无抑制活性。化合物3b与吉西他滨Gem表现出相似的肿瘤细胞抑制活性。化合物3a在MiaPaCa2, SW1990, MCF-7中均表现出较Gem更强的肿瘤细胞抑制活性。统计学分析(T-test)证实两组结构差异显著,P值分别为0.025, 0.044和0.035(图1)。
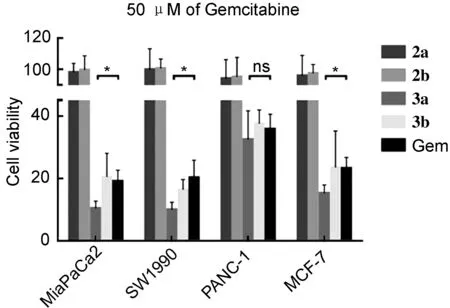
图1 目标化合物对4种肿瘤细胞的抑制活性Figure 1 Biological activities of target compounds on the four tumor cells
由图1还可知,3a药物组中MiaPaCa2, SW1990, PANC-1, MCF-7等4种细胞的活力分别为(10.35±1.34)%, (9.91±1.37)%, (32.41±5.33)%, (15.22±1.53)%,而对照组的4种细胞的活力分别为(19.01±2.08)%, (20.13±3.24)%, (35.72±2.78)%, (23.20±2.02)%。在PANC-1细胞中,3a较Gem表现出更高的细胞抑制率,但T-test结果显示差异不显著(P=0.61)。
此外,还测得3a对4种肿瘤细胞的IC50值均小于Gem组(表1)。而3b也显现出与原药吉西他滨相当的抗肿瘤活性。该实验结果表明,模型药物Gem在N4引入亲脂性功能基团以后,有望保持或更好地发挥其抑制肿瘤细胞增殖的活性。
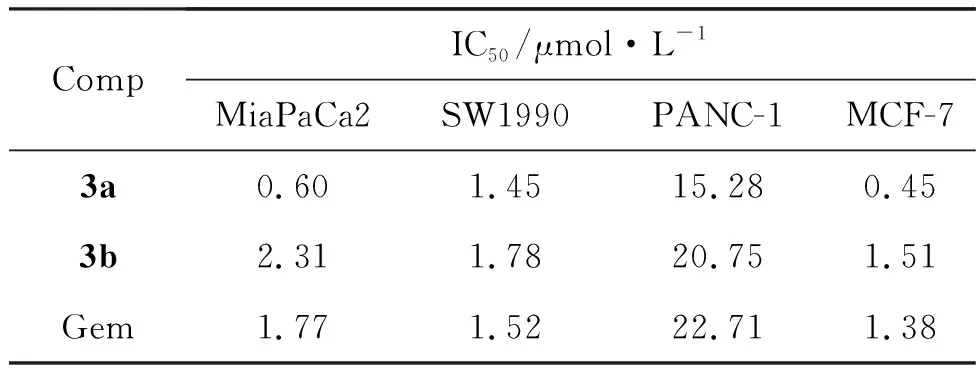
表1 化合物的体外抗肿瘤活性Table 1 In vitro antitumor activities of the compounds
2.2 代谢稳定性
文献[3]报道吉西他滨临床应用面临最严重的挑战是其极易被血液中大量存在的CDA氧化失活,导致代谢稳定性差。为探究3a表现出高抗肿瘤活性的原因,选择3a和吉西他滨测定了CDA稳定性(图2)。由图2可见,3a的吸光度与CDA无关联,而吉西他滨在CDA存在条件下的吸光度显著低于无酶条件下的吸光度,证明吉西他滨易被CDA还原而不稳定,而3a则不受CDA影响,表现出更好的稳定性。

Time/min图2 Gem和3a对CDA的稳定性Figure 2 The stabilities of Gem and 3a in the presence of CDA
借助羧酸类化合物,合成了具备高代谢稳定性的脂溶性吉西他滨衍生物(2a, 2b和3a, 3b)。体外抗肿瘤实验表明,3a和3b具有抗肿瘤活性,尤其3a不仅在多个细胞系上显现出强于吉西他滨的抗肿瘤细胞增殖活性,而且能对抗脱氧胞苷脱氨酶的作用,有望作为Gem的脂溶性前药进行深度开发。
猜你喜欢
世界农药(2022年10期)2022-11-10
炼油技术与工程(2022年8期)2022-08-18
浙江医学(2022年10期)2022-06-11
世界科学技术-中医药现代化(2022年2期)2022-05-25
医学信息(2022年8期)2022-04-23
能源化工(2021年2期)2021-12-30
昆明医科大学学报(2021年12期)2021-12-30
世界最新医学信息文摘(2021年12期)2021-06-09
临床医药实践(2021年4期)2021-04-10
农药科学与管理(2021年2期)2021-03-16
