
Prediction of fibril formation by early-stage amyloid peptide aggregation
2020-05-10 08:17JiaojiaoHuHuiyongSunHaipingHaoQiulingZheng
Jiaojiao Hu , Huiyong Sun , Haiping Hao , Qiuling Zheng ,**a
b Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Tongjiaxiang #24, Nanjing, Jiangsu, 210009, China
c Key Laboratory of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University, Tongjiaxiang #24, Nanjing, Jiangsu, 210009, China
d State Key Laboratory of Natural Medicines, China Pharmaceutical University, Tongjiaxiang #24, Nanjing, Jiangsu, 210009, China
Keywords:
ABSTRACTAmyloid fibrils are found in systemic amyloidosis diseases such as Alzheimer's disease, Parkinson's disease,and type II diabetes.Currently,these diseases are diagnosed by observation of fibrils or plaques,which is an ineffective method for early diagnosis and treatment of disease.The goal of this study was to develop a simple and quick method to predict the possibility and speed of fibril formation before its occurrence. Oligomers generated from seven representative peptide segments were first isolated and detected by ion-mobility mass spectrometry (IM-MS). Then, their assemblies were disrupted using formic acid (FA). Interestingly, oligomers that showed small ion intensity changes upon FA addition had rapid fibril formation.By contrast,oligomers that had large ion intensity changes generated fibrils slowly.Two control peptides(aggregation/no fibrils and no aggregation/no fibrils)did not show changes in their ion intensities,which confirmed the ability of this method to predict amyloid formation.In summary,the developed method correlated MS intensity ratio changes of peptide oligomers on FA addition with their amyloid propensities. This method will be useful for monitoring peptide/protein aggregation behavior and essential for their mechanism studies.
1. Introduction
Aggregation of proteins or peptides into amyloids is a recognized hallmark of various systemic amyloidosis-related diseases,including Alzheimer's disease, Parkinson's disease, and type II diabetes [1-3]. The formation of amyloids usually involves earlystage oligomerization and nucleation before growth of mature fibrils. Current diagnosis of these diseases is based on the observation of mature fibrils or plaques. Oligomer cytotoxicity, inhibitor screening, and conformational changes during oligomerization become popular areas of research, but the aggregation states of early-stage oligomers and their relationships with fibril formation have received less attention and remain unclear [4-8]. Recent studies have indicated that different oligomer forms could contribute to diverse pathways of mature fibril formation [9].Hence,understanding the aggregation behavior and propensity for amyloid formation of oligomers is critical for elucidating their pathological influences.
Conventional methods, including X-ray diffraction [10-12],transmission electron microscopy (TEM) [13-15], and solid-state nuclear magnetic resonance spectroscopy [16,17], are either limited for morphological characterization of mature fibrils, or providing averaged structural information only. Methods using thioflavin T(ThT)and Congo red are applied for kinetic monitoring;however,their detection mainly relies on the presence of abundant β-sheets or mature fibrils [18-22], and they are not suitable for early-stage oligomer characterization.
Mass spectrometry(MS)has been widely used for the analysis of protein and protein-protein interactions, including protein complex detection, kinetic studies [23], and conformational analysis
[24] by coupling with ambient ionization techniques, such as electrospray ionization (ESI) [25], desorption electrospray ionization (DESI) [26,27] and extractive electrospray ionization (EESI)[28,29].In the research field of amyloid protein,native MS enables the direct detection of oligomer species and with the assistance of ion mobility (IM), co-populated oligomers with different conformational states or n/z(n denotes the oligomerization number)can be resolved [30,31]. The applications of hydrogen/deuterium exchange (HDX) and chemical crosslinking (CX) coupling with MS allow the dynamic monitoring of conformational characterization of interacted sites involved in the onset of aggregation [32,33].Despite these advances, it remains difficult to determine the relevance of the assembly properties of early-stage oligomers and fibril formation.
Herein, we developed an ESI-MS strategy for simple and quick prediction of the amyloid propensity. Oligomers generated from seven representative peptide segments(Table S1)were selected to test the feasibility of the method. The generated oligomers were ionized by ESI and pulsed into an IM cell for separation.Formic acid(FA) was subsequently applied to disrupt the assemblies of the formed oligomers.Oligomers generated from each peptide showed characteristic ion intensity changes, which were used to differentiate their fibril growth properties. The results showed that oligomers with small ion intensity changes upon FA addition were correlated with a rapid fibril formation.By contrast,oligomers that generated fibrils slowly showed large ion intensity changes. TEM results confirmed that oligomers with a large range of ion intensity changes spent more time generating fibril than those with a small range of ion intensity changes. Additionally, no ion intensity changes were observed for the two control peptides (aggregation/no fibrils and no aggregation/no fibrils), which supported the ability of the developed method to predict amyloid formation.This approach will be useful for studies of amyloid formation mechanisms and assembly pathway analysis.
2. Materials and methods
2.1. Chemicals
All peptides were synthesized by Nanjing Peptide Biotech Ltd.(Nanjing, China) and were >98% pure. No further purification was required before IM-MS analysis. All other reagents (high-performance liquid chromatography-grade methanol, acetonitrile, acetic acid and FA) were purchased from Merck (Darmstadt, Germany).Deionized water was prepared using a Milli-Q system (Millipore,Billerica, MA).
2.2. Prefibril species preparation
Peptides were dissolved in MeOH/H2O (1:1, v/v) to a final concentration of 2 mM,and incubated at room temperature for 15 h to initiate generation of oligomers. The resulting solutions were centrifuged at 10,000×gfor 10 min before MS analysis.
2.3. IM-MS analysis
Data acquisition was performed on a Waters Synapt G2 Si Q-TOF(Milford, USA) in the positive ion mode. The following MS conditions were applied: capillary voltage, 2.5 kV; desolvation temperature,400°C;source temperature,120°C;cone gas flow rate,50 L/h; desolvation gas flow rate, 800 L/h; and nebulizer gas pressure,6.8 bar. For mobility mode, the following settings were used: helium cell gas flow rate,180 L/h;IMS gas flow rate,90 mL/min;IMS wave velocity,600 m/s;and IMS wave height,30 V.The mass range was set tom/z200-3000.Data were processed using MassLynx 4.1 software. The intensity ratio of each oligomer was calculated according to Eq. (1).

2.4. TEM analysis
An aliquot (8 μL) of the solution was dispersed on a 400-mesh microgrid and air dried before TEM (JEM-2100F, Tokyo, Japan) at 200 kV.
2.5. ThT analysis
Stock solutions(18 mM)were prepared in 1,1,1,3,3,3-hexafluoro-2-propanol and sonicated for 15 min to dissolve all aggregates.Aliquots of these stock solutions were dried and then dissolved in MeOH/H2O (1:1, v/v) containing 20 μM ThT to give a final concentration of 2 mM. Fluorescence spectra were recorded using a quartz cuvette with a 1 cm path length(Yixing Jingke,China)and a spectrofluorophotometer (FL-6500, PerkinElmer, Waltham, MA,USA). Spectra were recorded at room temperature from 460 to 500 nm after excitation at 440 nm (excitation slit width, 10 nm;emission slit width,10 nm). The fluorescence intensity at 480 nm was normalized using the value at 0 h.
2.6. Dynamics light scattering (DLS) analysis
The intensity-weighted mean hydrodynamic diameter distributions of freshly prepared VEALYL and NNQQNY, and oligomeric state VEALYL and NNQQNY after a 15 h incubation in MeOH/H2O(1:1,v/v)were determined by DLS using a Litesizer 500(Anton Paar,Graz,Austria).A scattering angle of 175°was used.Before analysis,the samples were first centrifuged for 5 min at 10,000×g. The supernatant from each sample was diluted using a solvent containing FA. The FA volume fractions were varied in different experiments.The data were analyzed using Kalliope software(Graz,Austria).The reported results were determined from the averages of at least three measurements.
3. Results and discussion
Three hexapeptides (VEALYL, NNQQNY, and SSTNVG) that reportedly form fibrils through different pathways were selected as samples(Table S1)[6].After incubation for 15 h to initiate oligomer formation (Materials and Methods), the three peptide solutions were tested by the ThT assay at an excitation wavelength of 440 nm.The fluorescence changes were negligible,which indicated that an incubation time of 15 h was not sufficient to generate saturated βsheet to be monitored by the ThT assay(Fig. S1).
The MS spectra (Fig. 1) showed coexistence of oligomers with broad n ranges,most of which could be assigned according to theirm/zvalues and isotopic distributions. In some cases, a singlem/zvalue, such asm/z1560.4 orm/z1950.0 (Fig. 1B), was consistent with multiple n/z theoretically and could not be well-resolved. In these cases,IM was employed to further isolate the oligomers with identicalm/zvalues (Fig. S2). On this basis, the hexapeptides formed oligomers with the following n/z values:VEALYL,up to 13/4; NNQQNY, up to 14/5; and SSTNVG, up to 15/4 (Table S2).
Next,we investigated the aggregate stability by adding FA to the ESI spray solvent.The volume fraction of FA ranged from 0.001%to 0.07%and the pH range was measured from 2.6 to 3.8,which could be considered negligible to influence the ionization efficiency.The relative intensity ratio of each oligomer was calculated according to Eq.(1)and plotted in Fig.2,respectively.For the oligomer species of VEALYL generated atm/z2297.6 with an n/z value of 13/4, the intensity decreased to 25%of the original MS intensity on addition of FA (Fig. 2A). By contrast, the singly charged monomer (n/z = 1/1)showed almost no change in intensity with FA addition.The other two peptides (NNQQNY and SSTNVG) also showed intensity decreases (Figs. 2B and C). Although MS intensity changes were observed for all of the oligomers, the extent of the decrease was different for each peptide. The MS intensity change for each oligomer was converted to a fold change using the ratio of maximum to minimum MS intensities (Fig. 3). The fold change of oligomers generated from VEALYL had a smaller range (3.6-5.3) compared with the other two peptides (NNQQNY, 8.3-15.6; and SSTNVG,7.2-23.5) (Table S3). The same analysis was carried out with another MS compatible acid, acetic acid, by using VEALYL and NNQQNYas examples.Similarly,VEALYL had a smaller range of fold change (1.6-2.2) compared with that of NNQQNY (1.1-2.2)(Fig.S3).Acetic acid trials had similar results to those obtained from FA, but the latter ones showed more significant differentiation,which could be explained by weak acidity of acetic acid compared with that of FA.Therefore,FA was selected all and after.In addition,in order to validate the intensity changes are amyloid propensity related,and to differentiate artificial oligomers due to ESI process or high concentration, two control peptides with different aggregation behaviors were selected, including VELYAL (mutant from VEALYL, aggregation/no fibrils) and PPTNVG (fragment of rat amylin, no aggregation/no fibrils) [34-36]. FA showed negligible effects on the oligomers generated by both peptides (Figs. S4-S6)as the fold changes were 1.1-1.4 for VELYAL and 1.7-1.8 for PPTNVG(Fig. 3). Thus, we propose that the fold change is amyloiddependent and can be used for prediction.

Fig.1. MS spectra of peptide (A) VEALYL, (B) NNQQNY, and (C) SSTNVG.
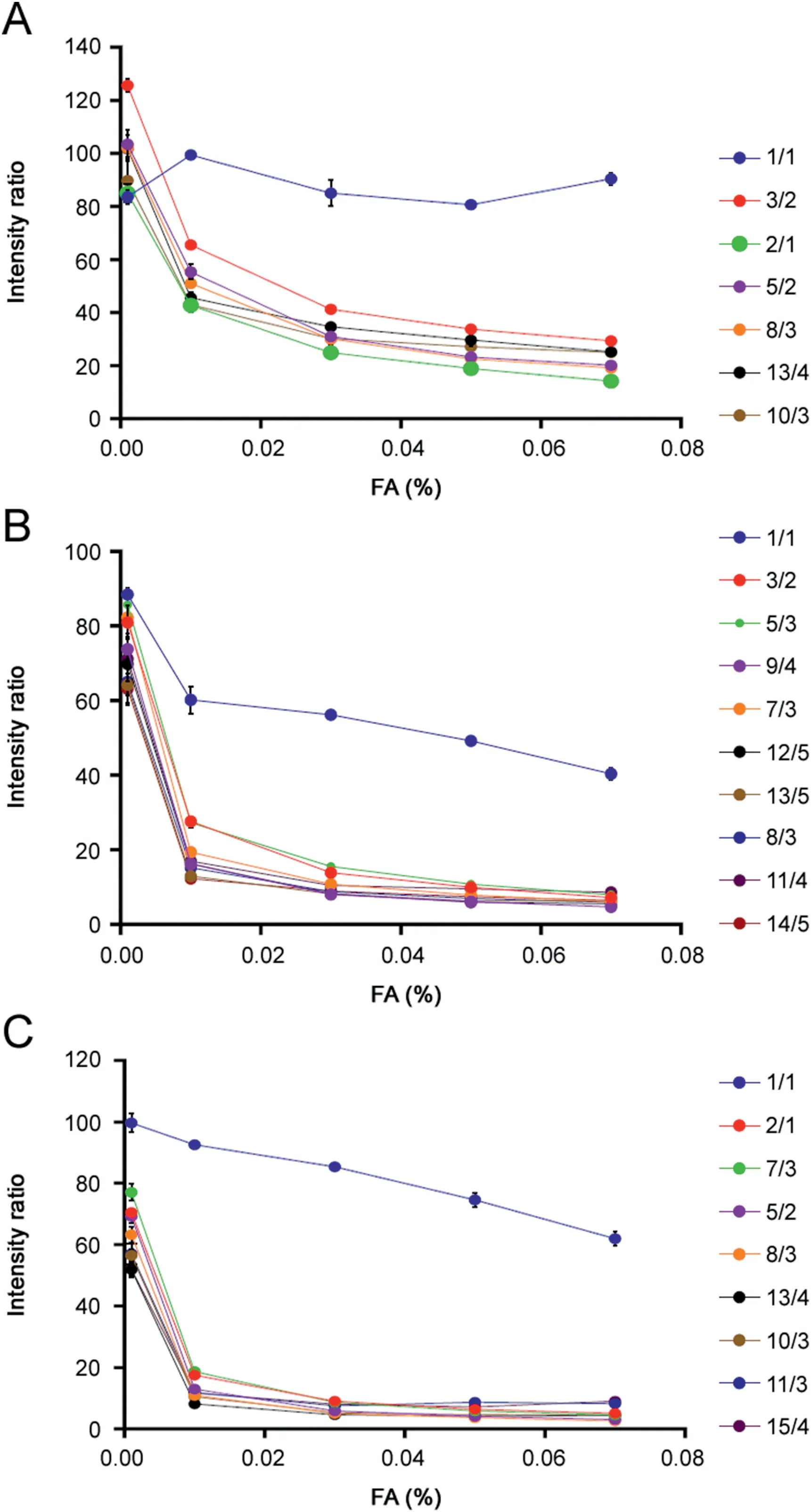
Fig. 2. MS ion intensity ratio of (A) VEALYL, (B) NNQQNY, and (C) SSTNVG. Data are presented as means ± SEM. Each data point was calculated according to Eq. (1) and represented the intensity ratio of a single oligomer between MS intensity obtained with different volumes of FA and without FA, respectively.
The oligomer degradation by FA was further confirmed by DLS experiments. After incubation for 15 h, the soluble particle sizes of both peptides VEALYL and NNQQNY were in the range 100-1000 nm.Upon dilutionwithsolvents containing different concentrationsofFA,the soluble particle size decrement of VEALYL was not that significant compared with that of NNQQNY, which decreased to 100-360 nm(Fig.4).Thisindicated thataddition of FA resulted indecomposition of theoligomers.Itwasalsoinlinewithpreviousfoldchangeresultsthat showed the particle size of VEALYL was affected less than that of NNQQNY, which suggested that the NNQQNY would be impacted more than VEALYL by FA addition.
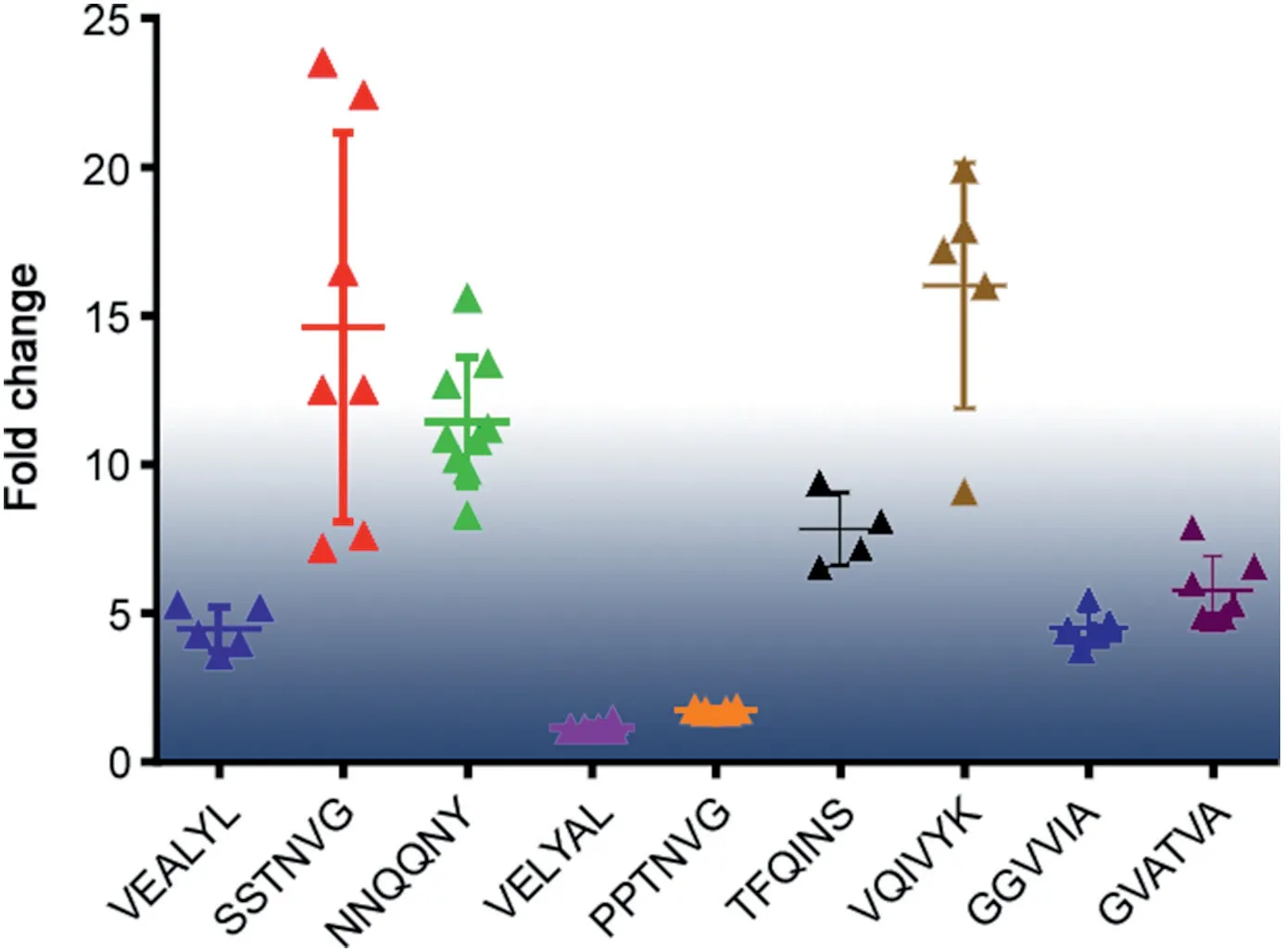
Fig. 3. Fold change of each oligomer generated from peptide VEALYL, SSTNVG,NNQQNY, VELYAL, PPTNVG, TFQINS, VQIVYK, GGVVIA, and GVATVA.

Fig. 4. DLS results for oligomers with 15 h incubation and diluted by (A) and (D) pure water; (B) and (E) 0.1% FA(v/v); and (C) and (F) 1% FA. Data are presented as means ± SD.
TEM was used to explore fibril growth after a 10 day incubation(details found in Materials and Methods). The TEM results (Fig. 5)showed that although the three peptides were amyloid peptides,a 10 day incubation was sufficient for VEALYL to develop obvious fibrils compared with NNQQNY and SSTNVG. According to the literature, VEALYL aggregation occurs via fibrillation through a single fibril strand to steric zipper pathway,whereas NNQQNY and SSTNVG fibrillation occurs via an intermediate state with disordered aggregation patterns[6].Aligned with our results,oligomers with small fold change ranges (e.g., VEALYL in Fig. 3) would be in relatively stable assemblies and likely to rapidly form fibrils. By contrast, oligomers with large fold change ranges (e.g., NNQQNY and SSTNVG in Fig.3)would be in unstable assemblies and undergo a long growth phase of hybrid aggregation before fibril formation.

Fig. 5. TEM images of peptide (A) VEALYL, (B) NNQQNY, and (C) SSTNVG. Scale bar:1.0 μm.
To validate the regularity of the correlation between MS ion intensity change and fibril formation, we tested four more peptides: TFQINS, VQIVYK, GGVVIA, and GVATVA (Table S1). After incubation to initiate oligomer generation,the analytes were ionized by ESI,which was followed by IM separation to match the n/z with theirm/zvalues(Table S2).According to them/zvalues from the MS spectra(Fig.S7)and drift times obtained from the IM(Fig.S8),the n/z values for the oligomers were up to 13/4 for TFQINS,13/4 for VQIVYK, 14/3 for GGVVIA, and 14/3 for GVATVA. Next, an FA gradient was applied to determine the MS intensity changes(Fig. S9), which were then converted to fold changes (Fig. 3). The fold change ranges were 6.6-9.4 for TFQINS, 3.8-4.7 for GGVVIA,and 4.8-7.9 for GVATVA(Table S3).These fold changes were small,which suggested the oligomer assemblies were stable and formed mature fibrils in less time than other oligomers. By contrast, oligomers from the peptide VQIVYK displayed a large fold change(9.1-19.9, Table S3), which indicated that they were unstable and would take more time to form fibrils. This was confirmed by TEM analysis after a 10 day incubation(Fig.S10),where TFQINS,GGVVIA,and GVATVA clearly showed generation of fibrils, but no obvious fibrils were generated for the peptide VQIVYK.
4. Conclusions
A widely applicable approach was developed to correlate aggregate stability with fibril formation according to the MS intensity changes of early-stage oligomers.The observed changes are representative of the different oligomer aggregation properties,which are consistent with their disparate aggregation patterns.The results suggest that oligomers with small ranges of fold change have ordered and homogenous aggregation patterns and form mature fibrils rapidly. Oligomers with large ranges of fold change undergo hybrid aggregation patterns in a relatively slow aggregation process. It is probably the different sizes or structures of oligomers formed by different peptides that cause the different ranges of fold change and further investigation is required to uncover the exact mechanism for the future work.Because of the sensitivity and speed of IM-MS, this approach will be helpful for mechanistic studies of aggregation and prediction of the ultimate fate of aggregates from early-stage oligomers. Moreover, the capacity for detecting and deducing oligomer assembly patterns at an early stage by this approach will be beneficial for understanding their aggregation behaviors and mechanism studies.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant Nos. 81930109, 81720108032, 81430091,81421005, 81703471, and 81603031), the Natural Science Foundation of Jiangsu Province (Grant No. BK20170740), the 111 project(Grant No. G20582017001), projects for Major New Drug Innovation and Development (Grant Nos. 2018ZX09711001-002-003 and 2018ZX09711002-001-004), the State Key Laboratory of Natural Medicines at China Pharmaceutical University, China (Grant No.SKLNMZZCX201817), and a “Double-First Rate” project (Grant No.CPU2018GF09).
We thank Prof. Weixing Xia and graduate student Ke Pei from the Ningbo Institute of Materials Technology and Engineering,Chinese Academy of Sciences, for the preparation of TEM samples and TEM analysis; and Prof. Wei Chen from the Department of Pharmaceutical Engineering, School of Engineering, China Pharmaceutical University, for DLS analysis.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2019.12.002.
 Journal of Pharmaceutical Analysis2020年2期
Journal of Pharmaceutical Analysis2020年2期
- Journal of Pharmaceutical Analysis的其它文章
- Recent advances and perspectives of nucleic acid detection for coronavirus
- Molecular immune pathogenesis and diagnosis of COVID-19
- Hollow fiber-based liquid phase microextraction followed by analytical instrumental techniques for quantitative analysis of heavy metal ions and pharmaceuticals
- Quantitative computed tomography analysis for stratifying the severity of Coronavirus Disease 2019
- Erucic acid from Isatis indigotica Fort. suppresses influenza A virus replication and inflammation in vitro and in vivo through modulation of NF-κB and p38 MAPK pathway
- Metabolic profiling of four synthetic stimulants, including the novel indanyl-cathinone 5-PPDi, after human hepatocyte incubation