
3D卷积神经网络的结构优化及中枢神经系统药物的识别
2020-04-29 00:44张瑞林丁彦蕊
西北大学学报(自然科学版) 2020年1期
张瑞林 丁彦蕊


摘要:该文研究了如何利用3D-CNN识别中枢神经系统(CNS)药物和non-CNS药物。首先,构建CNS药物和non-CNS药物数据集并优化小分子构象;然后,以3D网格矩阵编码小分子结构,作为3D-CNN模型的输入;接着,在模型训练中,采用正交实验法对3D-CNN模型的超参数进行快速优化;最后,使用外部测试集检验模型,达到ACC 为84.3%,MCC为0.685,AUC为0.884的泛化性能。实验表明,在正交实验法获取可靠超参数组合的基础上,3D-CNN模型对于CNS药物的识别具有良好效果,所构建的模型为设计新的CNS药物提供了基础。
关 键 词:3D卷积神经网络;药物虚拟筛选;正交实验
中图分类号:TP399
DOI:10.16152/j.cnki.xdxbzr.2020-01-005開放科学(资源服务)标识码(OSID):
Structural optimization of 3D convolutional neural network anddrug identification in central nervous system
ZHANG Ruilin1,2, DING Yanrui1,3
(1.School of Science, Jiangnan University, Wuxi 214122, China;
2.Jiangsu Key Laboratory of Media Design and Software Technology, Jiangnan University, Wuxi 214122, China;
3.Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China)
Abstract: How to identify the central nervous system (CNS) drug and non-CNS drug is studied in this paper. Firstly, the data set of CNS and non-CNS drugs were constructed and the conformations of molecules were optimized. Then, small molecule structures were encoded by 3D grid matrix as input of 3D-CNN model. In process of training model, orthogonal experiment method was used to optimize the hyper-parameters of 3D-CNN model quickly.Finally, external test sets were used to test the model, which achieved the generalization performance with ACC of 84.3%, MCC of 0.685 and AUC of 0.884. The experiments show that the 3D-CNN model has a good performance on the identification of CNS drugs based on obtaining reliable hyper-parameters combination by orthogonal experiment. The model constructed provides a basis for the design of new CNS drugs.
Key words: 3D convolutional neural network; drug virtual screening; orthogonal experiment
神经系统疾病使全世界数亿的患者饱受疾病的折磨,由于尚不完全了解其发病机制,所以致残率和死亡率一直居高不下[1-2]。其中,中枢神经系统(central nervous system,CNS)疾病在人类精神及行为方面造成了极大影响,由于人类大脑的自我保护机制,这类药物在进入大脑和脊髓时会遇到各种不同的障碍,使得新开发出来的候选药物大多很难到达CNS[3-4]。现有治疗CNS疾病的药物小分子在某些方面具有共同的性质,与其他药物小分子的性质差异决定了它们是否能够到达CNS,利用这种差异来筛选目标化合物是研究和开发有效的CNS药物的一个重要途径。
机器学习是目前最热门的学科之一,并在各个领域得到广泛应用,其特定的算法模型有些已经达到甚至超越人类的效果[5]。浅层机器学习早已应用在化合物识别和分子间作用力预测等方面[6-7],Cai等人以优化后的分子特征和拓扑指纹为描述符,构建朴素贝叶斯模型和基于描述符不同组合的递归划分模型,对二肽基肽酶IV抑制剂具有良好的预测能力[8];Ahamed等人利用E-Dragon,Ochem,PowerMV在组合数据库中提取的分子描述符,构建KNN模型对5-脂氧合酶蛋白抑制剂的预测准确率达到了77.9%[9];Lee等人以ECFP_4指纹图谱作为模型输入,利用随机森林方法构建结构活性关系,能够准确预测配体针对每个靶点的活性[10]。这些方法主要将计算的描述符数值作为模型输入,尽管其计算结果基本令人满意,但这种预先设计的描述符涉及大量的人为主观因素,而且训练模型前一般都要进行复杂耗时的特征选择操作。
随着大数据时代的到来,生物数据呈指数增加,而且药物小分子是一种极其复杂的结构,基于描述符的普通浅层学习无法学习到更深层次的特征信息[11-12]。近几年,以卷积神经网络(CNN)为代表的深度学习在图片识别上表现突出,这使其走进了药物发现研究人员的视野,特定的算法与框架使模型能够学习到更加深层次、更抽象的特征信息[13-14]。Kristina等人将深度学习应用于药物协同预测,利用化学与基因组信息作为模型输入,AUC性能达到了0.9[15]。分子指纹作为输入的深度学习模型不仅可以预测药物不良反应,还可以识别这些不良反应分子的子结构,帮助开展药物安全性研究[16]。基于权威的公共数据集,Andreas等人开发了DeepTox管道,在1.2万种化学品和药物的数据集上获得了比其他机器学习算法更好的毒性预测结果[17]。由于现实中的药物是基于三维空间的一种结构,基于3D卷积神经网络(3D-CNN)的方法被提出,用于给蛋白质配体结合构象打分[18],这种三维输入的方式避免了人为计算描述符的限制,保留了分子的最客观、最全面的信息。
尽管各种机器学习方法在药物识别方面得到广泛应用,但针对CNS药物的识别研究却很少。Jiang等人利用SVM建立了发现潜在的中枢神经系统化合物的机器学习模型,采用网格搜索、遗传算法和粒子群优化这3种参数优化方法对SVM模型进行优化,并对CNS化合物进行血脑屏障研究,为CNS药物的开发提供了一定参考[19]。Doniger等人利用多层感知器神经网络和SVM两种机器学习方法来构建不同分子的血脑屏障通透性模型,结果表明,SVM的性能优于神经网络,基于30多个不同的验证集,SVM可以准确预测96%以上的分子,30多个测试集的平均预测率只有81.5%[20]。Gao等人使用已知脑血稳态浓度比的药物分子来构建多项式核SVM模型,并用蒙特卡罗交叉验证获得了76.0%的准确率、0.739的AUC和0.760的F1评分,在SIDER数据库中成功筛选了110种可能穿透血脑屏障的药物[21]。这些方法都偏向于构建与血脑屏障相关的浅层机器学习模型,但不能保证能够穿越血脑屏障的小分子就一定是CNS药物,而且模型缺乏对更多的non-CNS药物小分子的学习。
本文首先构建了CNS药物与non-CNS药物数据集;然后,利用3D网格表示出优化后的药物小分子数据,并将其作为3D-CNN的输入;接着,利用正交实验法来设计模型的多个超参数组合,并确定最优的超参数组合;最终,建立可靠的用于识别CNS药物小分子的3D-CNN模型。
1 材料与方法
1.1 数据集的构建
ATC(anatomical therapeutic chemical)是由世界卫生组织开发的权威药物分类系统,根据药物的药理、化学等方面信息将药物分类[22-23]。本文在ZINC15数据库的基础上,统计基于ATC分类系统的药物小分子数据,并结合DrugBank数据库中对应药物的详细说明,收集治疗CNS疾病及non-CNS疾病的药物小分子数据。为了数据集的可靠性,排除一些重复的、含有金属原子的及包含多个小分子的药物数据,最终挑选出CNS药物小分子273个,non-CNS药物小分子879个。
所有药物小分子数据都是从ZINC15数据库中下载的SDF格式文件,这种格式包含了小分子中所有原子的基本信息及初始的三维坐标。为了保证所有小分子数据具有统一环境的最优三维构象,使用RDKit软件[24]对所有小分子数据进行基于MMFF94力场[25]的能量最小化。
1.2 药物小分子的3D表示
在识别图片时,CNN的输入是图片像素值的二维矩阵,然而小分子数据是空间的立体结构,重要的信息不仅包含原子的相对空间位置,还有每个原子的类型、所属子结构及原子之间的键类型等,药物小分子的表示是一个复杂的过程。本文采用3D网格表示法,将小分子放在一个3D网格中,以当前小分子中所有原子坐标的平均中心作为网格中心,网格大小默认为0.024 nm3,每侧的密度为0.05 nm,所以每个分子可以表示成一个48×48×48的三维矩阵。每个小网格中都有一个表示此处信息的属性值,用A(d,r)表示,A为此处受到周围重原子影响的大小,d表示此处与其他重原子中心的距离,r表示其他重原子的范德华半径,如式(1)所示。
A(d,r)=e2d2r2, 0≤d<r
4e2r2d2-12e2rd+9e2, r≤d<1.5r
0, d≥1.5r (1)
由于药物分子中重原子不止一种类型,为了区分不同原子所带来的不同信息,将不同原子类型放在不同通道,类似于图像的RGB三通道,根据smina[26]配体原子类型,本文采用18通道。图1为分子在3D网格中的示意图,圆球表示重原子,其中:灰色代表碳原子;紅色代表氧原子;蓝色代表氮原子;黄色代表硫原子。不同类型的原子存在不同的通道,不存在的类型以0补齐。
1.3 模型与训练
本文使用3D-CNN来训练识别CNS药物及non-CNS药物的深度学习模型,它与普通的图像识别模型类似,主要区别在于将二维矩阵的计算上升到对三维矩阵的计算。3D-CNN的优势在于样本的特征定义可以不受人为干扰,因为它可以自动学习到更深层次、更抽象的特征信息。以卷积层和采样层交替组成的特征提取层是3D-CNN的关键部分,卷积层负责对不同特征组合的抽象,采样层使最重要的特征得以保留。一般每个卷积部分后面会有一个激活函数,用于增强网络的表达能力,然后,通过全连接层将卷积部分提取的特征信息与真实标签进行映射,训练好的网络通过输出层输出预测结果。
由于3D-CNN算法的复杂性,合理的超参数选择是获得可靠模型的基础。基于Caffe深度学习框架[27]及Gnina软件[18],使用小批量样本训练网络,批大小为64,采用随机梯度下降法作为优化算法,最大迭代次数设置为20 000。由于分子数据的复杂性,提高初始学习速率,设置为0.1,动量设置为0.9,学习策略设置为inv,学习速率会随着迭代的进行逐渐变小,这可以提高算法寻优能力。
在构建的数据集中,CNS药物比non-CNS药物少很多,这里采用向下采样的方法来平衡数据集,最终选定CNS药物273个,non-CNS药物355个。从正负样本中分别随机选择25%的数据作为外部测试集,其余的作为交叉验证集。在交叉验证过程中,将交叉验证集随机分成5份,每次随机取其中4份用于训练模型,剩余的用来验证模型,重复这个过程5次。
1.4 正交实验法
正交实验是一种研究多因素多水平的设计方法[28-29]。3D-CNN结构中有许多超参数,共同决定着模型的训练,为了确定最佳的超参数组合,本文挑选了4个重要且相互独立的超参数:卷积核大小、卷积步长、下采样核大小及步长和初始卷积核个数,正交表如表1所示,L9(34)代表着考察4个因素,每个因素依据经验设置3个水平值,一共9种不同超参数组合。在以往的经验中,单独的卷积核大小对结果没有绝对性的影响,一般取奇数;卷积步长越小学习得越仔细,但训练时间也随之增长或导致过学习;下采样方式采用最大不重复采样,所以核大小与步长保持一致;卷积核个数会随着卷积层的递进而增加,平衡了下采样时放弃的不重要信息。我们发现,单个卷积层时的训练难以收敛,并且第4个卷积层时的样本矩阵缩为单个数值而缺乏卷积效果,所以,分别在只有两个和三个卷积层的情况下进行正交实验,最终确定最佳超参数组合。
1.5 模型评价指标
深度学习模型是一种复杂的结构,为了评估模型的性能,本文统计了真阳性(true positive,TP)即正确预测正样本的数量;真阴性(true negative,TN)即正确预测负样本的数量;假阳性(false positive,FP)即错误预测成正样本的数量;假阴性(false negative,FN),即错误预测成负样本的数量。基于这些统计,计算常用的模型评价指标,如灵敏度SEN=TP/(TP+FN),即正样本预测率;特异性SPE=TN/(TN+FP),即负样本预测率;准确度ACC=(TP+TN)/(TP+TN+FP+FN),用来衡量总体预测率;马修相关系数MCC=(TP×TN-FP×FN)/[(TP+FP)(TP+FN)(TN+FP)(TN+FN)1/2],用来衡量模型可靠程度;接受者操作特性曲线下面积AUC则是另一个评价模型整体性能的标准,它表示真阳性与假阳性组成的点形成的曲线与x轴围成的面积。通过以上评价指标从不同方面评估构建的CNS药物识别模型,值越大表示模型的性能越好。
2 结果与分析
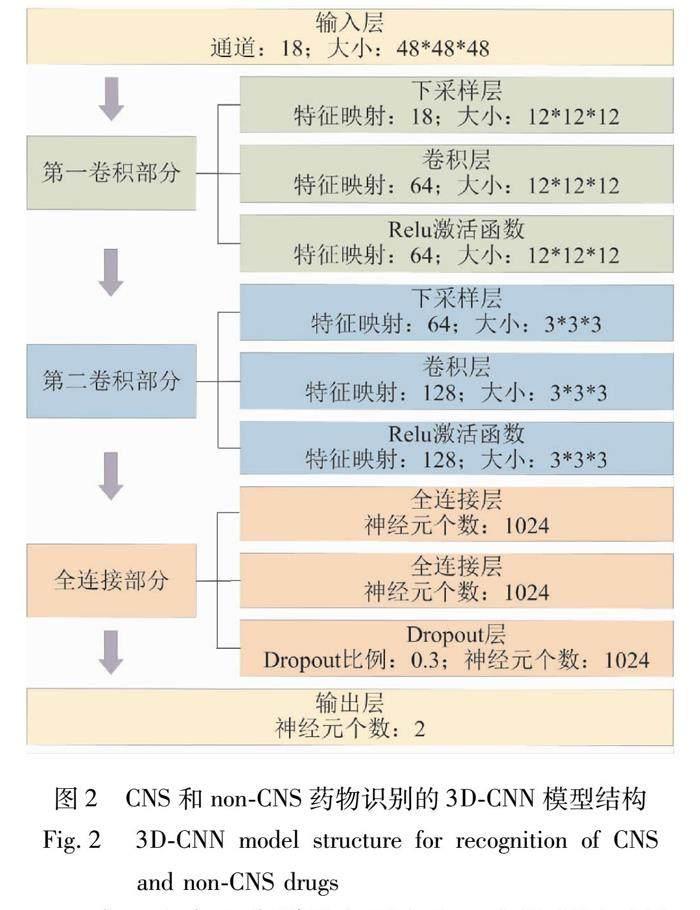
3D-CNN的多个超参数直接影响CNN的结构,本文根据正交实验进行参数组合,比较每种组合下模型的交叉验证结果,从而构建可靠的3D-CNN模型,模型结构如图2所示。
表2和表3分别是在两个和3个卷积层时的正交实验结果,其中,每个数值都是五折交叉验证的平均。使用3个卷积层时,最佳参数是第6个超参数组合,即卷积核大小为5,卷积步长3,下采样核大小及步长为2,初始卷积核个数为64,此时所有预测率都在80%以上,MCC和AUC分别为0.629,0.848。当采用两个卷积核时,最佳参数是第7个超参数组合,即卷积核大小为7,卷积步长1,下采样核大小及步长为4,初始卷积核个数为64,此时的SEN比3个卷积核时低0.5%,SPE基本不变,ACC,MCC,AUC也都略低于3个卷积核时的结果。然而,这两种结果非常接近,而且参数组合相同时,不同的卷积层数并没有表现出明显的结果差异,这与Simonyan[30]提到的两个3*3卷积层与一个5*5卷积层的效果几乎相同的情况一致,不同的卷积层数通过调整其他超参数也可以达到类似的效果。
为了进一步探索其他超参数对结果的影响,在上述两个最佳参数组合基础上,默认每个全连接层固定1 024个神经元,统计全连接层数为1,2,3时的结果,如表4所示。在两个卷积层后连接两个全连接层时,CNS药物和non-CNS药物预测率均为81%以上,ACC,MCC,AUC分别为82.7%,0.656,0.852,然而3个卷积层并没有达到这样的效果。全连接层将卷积部分输出的特征信息与各自标签进行映射,在只有一个全连接时,2Conv(7/1/4/64)+1FC与3Conv(5/3/2/64)+1FC的结果非常接近,在添加了两个全连接层后,3Conv(5/3/2/64)+2FC对药物信息提取的误差被放大,而2Conv(7/1/4/64)+2FC仍然很健壮,这是由于较大的卷积核与较小的卷积步长搭配时,使得更多的信息被抽象到不同层面,下采样层则保留了更多重要的信息。
尽管两个卷积层连接两个全连接层时的效果最好,这里仍然可能存在过拟合问题。Dropout是一种可以有效预防神经网络过拟合的方法[31]。由于每次迭代輸出层的结果影响着权重的修正,从而影响模型的训练,所以,本文在输出层之前添加Dropout层。如表5所示,在Dropout比例为0.3(0.3Dropout)时,ACC与MCC分别为82.3%,0.647,分别比不使用Dropout时低0.4%,0.09,但其AUC却高出0.007。这是由于使用0.3Dropout时,尽管预测正确的样本数量小于不使用Dropout,但模型对CNS药物的预测值更偏向于1,对non-CNS药物的预测值更偏向于0,即对每个药物的预测值更加接近真实值(1或0),致使AUC值稍微偏大。由于Dropout具有防止过拟合的作用,使用外部测试集来检测它的泛化能力,可以更加全面地评价一个模型的可靠程度。
为了验证上述的0.3Dropout是否增强了模型的鲁棒性,将不使用Dropout和使用0.3Dropout的模型分别对独立测试集进行测试。从图3中看到,使用0.3比例的Dropout层时,模型对独立测试集的泛化能力更好,SPE从81.5%上升到85.2%,ACC,MCC,AUC分别提升了1.9%,0.037,0.016。这表明,通过随机丢弃全连接层30%的神经元,增强了模型的健壮性,使模型在CNS药物与non-CNS药物的识别上具有更好的泛化能力。
3 結 语
随着3D-CNN在图像及视频识别上的快速发展,对现存的CNS及non-CNS药物小分子进行收集、整理和优化,并按其客观形状表示成代表自身的三维网格矩阵,构建基于小分子三维输入的3D-CNN深度学习模型,并基于正交实验法对模型结构进行优化,最终对CNS药物与non-CNS药物的识别达到了ACC为84.3%,MCC为0.685,AUC为0.884的预测性能。这表明基于正交实验法的3D-CNN模型在识别三维药物小分子上具有巨大潜力,这种技术的新应用将为相关药物研发人员提供有力的支持,并有希望加速未来的新药发现进程。
在实验过程中,正交实验法在不同独立参数搭配上起到了关键作用,其利用不同参数在不同水平值上的正交特性,找到了一种最接近最优情况的超参数组合,不仅为最优深度学习结构的确定提供了保障,还节省了大量的调参时间。但是,仍然存在一些问题,网格的大小及密度影响着小分子数据的矩阵表示,更小的密度会导致更复杂的分子矩阵,这要求更高的硬件条件。当前CNN应用大多是针对图像这样的二维数据,在这样的基础上,本文探索的针对三维卷积的卷积结构可能还存在提升空间,这需要考虑更多的因素。
参考文献:
[1] NI J, HAN F, YUAN J, et al. The discrepancy of neurological diseases between china and western countries in recent two decades[J].Chinese Medical Journal, 2018, 131(8): 886-891.
[2] KIM C J. Understanding of neurological diseases [J].International Neurourology Journal, 2018, 22:103.
[3] MISRA A, GANESH S, SHAHIWALA A, et al. Drug delivery to the central nervous system: A review [J].Journal of Pharmacy and Pharmaceutial Sciences,2003,6(2):252-273.
[4] SRINIVAS N, MAFFUID K, KASHUBA A D. Clinical pharmacokinetics and pharmacodynamics of drugs in the central nervous system [J].Clinical Pharmacokinetics, 2018, 57(9): 1059-1074.
[5] PALUSZEK M, THOMAS S. An overview of machine learning [M]∥MATLAB Machine Learning Recipes.Berkeley:Springer,2019:1-18.
[6] HOCHREITER S, KLAMBAUER G, RAREY M. Machine learning in drug discovery [J].Journal of Chemical Information and Modeling, 2018, 58(9): 1723-1724.
[7] LO Y C, RENSI S E, TORNG W, et al. Machine learning in chemoinformatics and drug discovery [J]. Drug Discovery Today, 2018, 23(8): 1538-1546.
[8] CAI J, LI C J, LIU Z H, et al. Predicting DPP-IV inhibitors with machine learning approaches [J].Journal of Computer-Aided Molecular Design, 2017, 31(4): 393-402.
[9] AHAMED T S, RAJAN V K, SABIRA K, et al. QSAR classification-based virtual screening followed by molecular docking studies for identification of potential inhibitors of 5-lipoxygenase [J].Computational Biology and Chemistry, 2018, 77:154-166.
[10]LEE K, LEE M, KIM D. Utilizing random forest QSAR models with optimized parameters for target identification and its application to target-fishing server [J]. BMC Bioinformatics, 2017, 18:75-86.
[11]CHEN X W, LIN X. Big data deep learning: Challenges and perspectives [J]. IEEE Access, 2014, 2:514-525.
[12]ZHANG L, TAN J J, HAN D, et al. From machine learning to deep learning:Progress in machine intelligence for rational drug discovery [J]. Drug Discovery Today, 2017, 22(11): 1680-1685.
[13]LECUN Y, BENGIO Y, HINTON G. Deep learning [J]. Nature, 2015, 521(7553): 436-444.
[14]CHEN H M, ENGKVIST O, WANG Y H, et al. The rise of deep learning in drug discovery [J]. Drug Discovery Today, 2018, 23(6): 1241-1250.
[15]PREUER K, LEWIS R P, HOCHREITER S, et al. DeepSynergy: Predicting anti-cancer drug synergy with deep learning [J]. Bioinformatics, 2018, 34(9): 1538-1546.
[16]DEY S, LUO H, FOKOUE A, et al. Predicting adverse drug reactions through interpretable deep learning framework [J]. BMC Bioinformatics, 2018, 19(21): 476.
[17]MAYR A, KLAMBAUER G, UNTERTHINER T, et al. DeepTox: Toxicity prediction using deep learning [J]. Frontiers in Environmental Science, 2016,3:80.
[18]RAGOZA M, HOCHULI J, IDROBO E, et al. Protein-ligand scoring with convolutional neural networks [J]. Journal of Chemical Information and Modeling, 2017, 57(4): 942-957.
[19]JIANG L D, CHEN J H, HE Y S, et al. A method to predict different mechanisms for blood-brain barrier permeability of CNS activity compounds in Chinese herbs using support vector machine [J]. Journal of Bioinformatics and Computational Biology, 2016,14(1):1650005.
[20]DONIGER S, HOFMANN T, YEH J. Predicting CNS permeability of drug molecules: Comparison of neural network and support vector machine algorithms [J].Journal of Computational Biology, 2002, 9(6): 849-864.
[21]GAO Z, CHEN Y, CAI X S, et al. Predict drug permeability to blood-brain-barrier from clinical phenotypes:Drug side effects and drug indications [J]. Bioinformatics, 2017, 33(6): 901-908.
[22]SKETRIS I S, METGE C J, ROSS J L, et al. The use of the world health organisation anatomical therapeutic chemical/defined daily dose methodology in Canada [J].Drug Information Journal, 2004, 38(1): 15-27.
[23]SKRBO A, BEGOVIC B, SKRBO S. Classification of drugs using the ATC system (anatomic, therapeutic, chemical classification) and the latest changes [J].Medicinski Arhiv, 2004, 58(1 Suppl 2):138-141.
[24]LANDRUM G. RDKit: Open-Source cheminformaticssoftware[EB/OL].2018,http://www.rdkit.org/.
[25]HALGREN T A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94 [J]. Journal of Computational Chemistry, 1996, 17(5/6): 490-519.
[26]KOES D R, BAUMGARTNER M P, CAMACHO C J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise [J]. Journal of Chemical Information and Modeling, 2013, 53(8): 1893-1904.
[27]JIA Y, SHELHAMER E, DONAHUE J, et al. Caffe:Convolutional architecture for fast feature embedding[C]∥Proceedings of the 22nd ACM International Conference on Multimedia. ACM,Florida,2014:675-678.
[28]GEORGIOU S D. Orthogonal designs for computer experiments [J]. Journal of Statistical Planning and Inference, 2011, 141(4): 1519-1525.
[29]周新宇, 吳志健, 王明文. 基于正交实验设计的人工蜂群算法[J]. 软件学报, 2015, 26(9): 2167-2190.
ZHOU X Y, WU Z J, WANG M W. Artificial bee colony algorithm based on orthogonal experimental design[J]. Journal of Software, 2015, 26(9): 2167-2190.
[30]SIMONYAN K, ZISSERMAN A. Very deep convolutional networks for large-scale image recognition [J]. Computer Science, 2014.
[31]SRIVASTAVA N, HINTON G, KRIZHEVSKY A, et al. Dropout: A simple way to prevent neural networks from overfitting [J].Journal of Machine Learning Research, 2014, 15(1): 1929-1958.
(编 辑 李 静)
猜你喜欢
成都信息工程大学学报(2021年5期)2021-12-30
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
北京航空航天大学学报(2021年9期)2021-11-02
电子制作(2019年11期)2019-07-04
北京航空航天大学学报(2018年1期)2018-04-20
河北科技大学学报(2015年5期)2015-03-11
电测与仪表(2014年2期)2014-04-04
电视技术(2014年19期)2014-03-11
