
葡萄糖碳化物/海泡石复合材料制备工艺及响应面法优化*
2020-04-28 10:24郑锡瀚马忻狄覃璐瑶张瑞莲蓝丽红
功能材料 2020年4期
郑锡瀚,马忻狄,覃璐瑶,张瑞莲,蓝丽红
(广西民族大学 化学化工学院,广西多糖材料与改性重点实验室,广西高校化学与生物转化过程新技术重点实验室,南宁 530006)
0 引 言
海泡石为含氧盐类矿物,其主要为金属元素阳离子与硅酸根结合的化合物,属层链结构纤维状含水的镁硅酸盐矿物[1]。天然海泡石以纤维状存在,内部空隙多且大,比表面积较大且吸附性能良好[2-3]。其理论化学式为Mg8[Si12O30](OH)4·12H2O,分子中含有较多的结晶水,这是影响海泡石吸附容量的重要因素[4]。因此,我国虽有丰富的海泡石矿产储量和开采量,但目前多数直接以原料廉价出口[5]。
近年来,如何通过物理和化学方法改性提高海泡石吸附性能成为矿物加工和应用领域的研究热点。李雪婷等[6]利用酸改性方法,制备出了对重金属有抑制作用以达到修复土壤效果的酸改性海泡石;张建生等[7]用腐植酸钠、陶瓷减水剂AST、碳酸钠、盐酸灯作为改性剂,对海泡石进行改性;黄阳等[8]利用海泡石、CuSO4、FeCl3合成出CuFe2O4/海泡石复合材料,并探究了复合材料对Cu2+的吸附性能;Ying Ma等[9]对海泡石进行有机改性,以海泡石纳米纤维为载体制成了核壳Ag@Pt纳米颗粒。
而在矿物材料表面采用多糖类物质碳化法制备多功能复合型材料因其反应条件温和可控、能耗低、经济绿色而被备受关注。针对葡萄糖[10-11]、果糖[12]、纤维素[13]、淀粉[14]等生物糖类物质的水热碳化反应的研究在国内外均有见报导。碳水化合物在较高温度和压力的条件下,会脱水成类似呋喃的分子,再进行聚合和碳化,形成的碳化微球[15]。生物糖类物质在经过水热碳化反应后会产生存在多种活性官能团(如,C—H、C=O和 C=C等)的不定性的碳化微球,使其更易与其他分子、离子及官能团结合的,使附着有多糖碳化物的矿石材料表面具有更强大的吸附功能,并且在极大程度上拓展了海泡石基复合材料的应用领域[16-17]。
因此,实验以海泡石和葡萄糖为原料,利用水热碳化法,制备葡萄糖碳化物/海泡石复合材料,并使用真空冷冻干燥器干燥复合样品。复合材料的表征手段选定为X射线衍射分析(XRD)、红外吸收光谱分析(IR)、扫描电镜(SEM)、比表面积仪(BET)。选择亚甲基蓝作为吸附质,以复合材料对海泡石的吸附量为性能考察标准,通过单因素和响应面法优化了材料制备的工艺。旨在获得一种吸附性能优良的复合型海泡石材料,提高海泡石的附价值的同时,又能为地区丰富的多糖类物质利用开辟新的应用途径,为海泡石和多糖类物质的深加工及应用提供实验依据和较好的技术指导。
1 实验部分
1.1 原料与仪器
试剂:海泡石(Aldrich),葡萄糖(国药集团),六水合硫酸亚铁铵(阿拉丁试剂),亚甲基蓝(Solarbio,含量为98%~103%,MB),以上试剂均为分析纯。
仪器:MAGNA-IR550型傅立叶变换红外光谱仪(美国尼高力仪器公司),SUPRA 55 Sapphire场发射扫描电子显微镜(德国卡尔蔡司公司),Rigaku miniflex 600 X-ray diffractometer(Tokyo, Japan),麦克ASAP2460(BET),TU-1810PC紫外可见光分光光度计。
2 实验方法
2.1 葡萄糖碳化物/海泡石复合材料制备
将4.5 g葡萄糖溶于蒸馏水中,再将0.5 %(质量分数)六水合硫酸亚铁铵(催化剂)加入葡萄糖溶液中,待溶解之后,按比例(葡萄糖与海泡石的添加质量比)添加海泡石,与35 mL 蒸馏水混合,配置成混合悬浊液。经过充分搅拌30 min之后形成悬浮液,再将悬浮液超声分散30 min,使反应体系充分分散混合均匀。最后转移混合样品于50 mL不锈钢反应釜的聚四氟乙烯内衬中(实验过程中保持样品量占釜内体积约为80%,主要为了控制各反应釜内部压强等条件一致),180 ℃下进行水热碳化反应8 h,所得样品使用无水乙醇与蒸馏水交替洗涤,直至肉眼观察下滤液为无色时,将粗样品进行真空冷冻干燥后,即可得到葡萄糖碳化物/海泡石复合材料。
2.2 葡萄糖碳化物/海泡石复合材料对亚甲基蓝的吸附
准确称取0.1020 g亚甲基蓝(简称MB,试验用亚甲基蓝含量为98%~103%),蒸馏水为基体溶剂,配制得到实验用的浓度为100 μg/mL的亚甲基蓝原液,之后逐级稀释,分别移取0、0.50、1.25、2.50、3.75 mL的亚甲基蓝溶液(100 μg/mL),可得到浓度分别为:0、1.0、2.5、5.0、7.5、10、12.5 μg/mL的系列浓度梯度的亚甲基蓝溶液,以蒸馏水作为参比,在662 nm处测定亚甲基蓝系列溶液的吸光度,并绘成标准曲线[18-19]。
移液管准确移取50 mL 亚甲基蓝原液于150 mL的具塞锥形瓶中,加入0.110 g复合材料样品,室温条件下振荡4 h吸附后,于10 000 r/min条件下离心分离,吸取上层清液进行吸光度值测定,并计算海泡石对亚甲基蓝的吸附量。
处理实验所得数据可得,亚甲基蓝吸光度值的标准曲线为:Y=0.205X+0.005,R2=0.9999。
即本次实验中,吸附量采用式(1)进行计算。

(1)
3 结果与讨论
3.1 材料表征
3.1.1 扫描电镜(SEM)
图1为天然海泡石(50K倍电镜下)、葡萄糖碳化物(5K倍电镜下)、葡萄糖碳化物/海泡石复合材料(50K倍电镜下)的SEM图。如图可发现:天然海泡石在电镜下呈现为疏松多孔的纤维束状形貌,且空隙较大;葡萄糖水热碳化物在电镜下为直径 5~9 μm的碳化微球;而从葡萄糖碳化物/海泡石复合材料的SEM图中发现,球形的葡萄糖碳化物存在于海泡石纤维表面,并且呈现非均匀分布,碳化微球的直径普遍变小(约为0.10~0.15 μm),且大小较为均匀,而形状却可保持球状,是海泡石独特的空间结构和作用力导致。

图1 3种材料的SEM图
3.1.2 傅里叶变换红外光谱仪(FT-IR)
图2为天然海泡石、葡萄糖碳化物/海泡石复合材料和碳化后葡萄糖(碳化微球)的红外光谱分析。由图2可看出,复合材料的红外特征吸收峰分布与原料海泡石更为相似。在3 683 cm-1处存在海泡石内部与镁离子相连的O—H的伸缩振动的吸收峰。而3 642~3 010 cm-1之间出现较宽的吸收带,这是海泡石内的Si—O结合水分子的H—O—H 以及吸附水分子 H—O—H 伸缩震动产生的吸收带。而1 208 cm-1处则为O—H的弯曲振动峰。在1 075~977 cm-1处表现出的较强的吸收带是Si—O—Si伸缩振动所导致的。
而复合材料与海泡石原料的红外谱图的区别之处在于,复合材料的谱图在2 979~2 924 cm-1处和1 453~1 334cm-1处分别表现出了饱和羟基的C—H吸收弯曲振动和伸缩振动的吸收。且在1 695和1 614 cm-1所出现的两处吸收峰,分别对应C=O和C=C的吸收峰。
再对比碳化后的葡萄糖的红外吸收谱图,在2 979~2 924 cm-1处、1 453~1 334cm-1处、1 695和1 614 cm-1处均有特征吸收表现,因此说明,复合材料总体的红外谱图以海泡石的谱图为骨架,并且碳化后葡萄糖的特征吸收区域在复合材料上有所吸收表现。

图2 海泡石、葡萄糖碳化物/海泡石复合材料和碳化后葡萄糖(葡萄糖碳化微球)红外光谱分析
3.1.3 X射线衍射仪(XRD)
图3是海泡石、葡萄糖碳化微球、葡萄糖碳化物/海泡石复合材料的XRD谱图。由图3可知,在材料复合前后,晶型基本无改变;与经碳化处理后的海泡石的XRD图谱相比较,葡萄糖碳化物/海泡石复合材料的XRD图谱的峰强度略有降低,除此之外,无峰偏移等明显变化出现;葡萄糖碳化微球的图谱在20~23°之间形成无定型碳的宽化衍射峰,与其比较后,发现在复合材料谱图中无形成无定形碳的宽化衍射峰。

图3 海泡石、葡萄糖碳化微球、葡萄糖碳化物/海泡石复合材料的XRD谱图
3.1.4 比表面积仪(BET)
从图4可以看出,海泡石与葡萄糖碳化物/海泡石复合材料的N2吸附-脱附等温线可以看出,皆属于含有滞后环的Ⅳ型吸脱附等温线,根据脱附曲线来算出孔径分布。因此,利用BET法计算得到海泡石与葡萄糖碳化物/海泡石海泡石复合材料的比表面积,用BJH法求孔径。最终计算得到葡萄糖碳化物/海泡石海泡石复合材料的比表面积为120.8770 m2/g,明显小于比表面积为181.5825 m2/g 的海泡石,这符合随着负载量的增加而比表面积逐渐变小的趋势。由图5的孔径分布情况,分析可得海泡石与葡萄糖碳化物/海泡石海泡石复合材料的孔径大部分发布在10~40 nm之间,用 BJH 法求得在脱附过程中的平均孔径为21.8019 nm。

图4 海泡石与葡萄糖碳化物/海泡石复合材料的N2吸附-脱附等温线

图5 海泡石与葡萄糖碳化物/海泡石海泡石复合材料的孔径分布情况
3.2 单因素实验优化葡萄糖碳化物/海泡石复合材料制备工艺
3.2.1 葡萄糖与海泡石质量比对复合材料吸附量的影响
图6是葡萄糖与海泡石质量比对复合材料吸附量的影响趋势曲线。由图6可见,当葡萄糖与海泡石质量比的提高,复合材料对亚甲基蓝的吸附量呈现先增高后下降的趋势。在质量比为0.5∶1.0~1.5∶1.0的条件范围内,吸附量趋势表现为增高,是因为葡萄糖会不断发生脱水反应,导致固相产物的碳元素含量提高,葡萄糖碳化微球数量增加,海泡石上负载的碳化微球量同样增加,吸附量随着提高。而在质量比为1.5∶1~3.0∶1的条件范围内,吸附量呈现降低趋势,且下降程度趋于平缓,这是由于反应釜中存在过量的葡萄糖,葡萄糖在脱水后会大量团聚,且会占据大量空间,堵塞海泡石的孔隙,导致吸附量降低;另一方面,葡萄糖量添加过多,导致反应釜中投料量过多,反应釜内部压力改变减少了碳化微球的活性基团的产生,影响葡萄糖的碳化效果,在负载于海泡石表面后,影响复合材料的吸附效果。因此当葡萄糖和海泡石质量比例为1.5∶1时最佳,此时吸附值为42.64 mg/g。
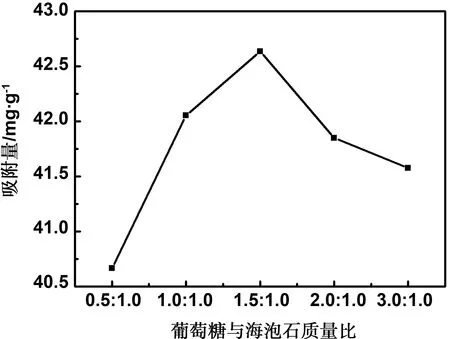
图6 葡萄糖与海泡石质量比对复合材料吸附量的影响趋势曲线
3.2.2 碳化时间对复合材料吸附量的影响
图7是碳化时间对复合材料吸附量的影响趋势曲线。由图7可见,材料碳化时间为8 h时,所得样品在本批实验样品中吸附值达到最大,当反应时间少于8 h时,碳化反应未开始或者样品未碳化完全,无法生成或生成的活性功能团的数量减少,导致所制成的复合材料吸附量减少。反应时间过长(即反应时间超过8 h)时,碳化反应所生成的焦油等多种副作用产物没有及时洗脱,在高温条件下,更容易被海泡石所吸附,堵塞海泡石孔隙,难以洗脱,因此,导致复合材料吸附性能下降。故将8 h定为最佳碳化时间,该温度条件下测得样品吸附值为44.55 mg/g。
(4) 结合地质资料及钻孔资料分析认为,研究区热储主要为三叠系砂、板岩地层与第四系地层中的构造裂隙及第四系的松散孔隙。地热水经深循环,自深部热储沿雅拉沟断裂上行,在上行过程中受构造裂隙影响,与冷水发生混合;上升至近地表后,在构造破碎带及第四系地层中形成次生热储。因此在进行热水钻探及开发利用地热资源的过程中,需避免钻孔打穿次生热储而出现没无热水的情况。

图7 碳化时间对复合材料吸附量的影响趋势曲线
3.2.3 碳化温度对复合材料吸附量的影响
图8反映的是碳化温度对复合材料吸附量的影响趋势曲线。由图8可见,随着材料的碳化温度的升高,葡萄糖碳化物/海泡石复合材料对亚甲基蓝的吸附量呈现先增高后下降的趋势。控制温度达到150~170 ℃的范围内,吸附量趋势表现为增高,是因为过低的温度会影响葡萄糖的碳化程度,导致葡萄糖碳化反应不完全,会影响了活性基团的产生,影响吸附量;且过低的温度条件下,会导致海泡石内部残留大量的结晶水无法逸出,占据吸附空间,从而减弱吸附性能。而随着温度的升高结晶水逐渐失去,复合材料孔隙更加通畅,吸附量逐渐提高。然而,当碳化温度从170 ℃升高到210 ℃的时候,所测的样品吸附量呈现逐渐减低的情况,是由于温度过高会导致副反应的生成的同时会导致反应的最初阶段,部分葡萄糖被水解或降解,完成碳化反应的原料供给受到影响,导致葡萄糖碳化量下降,从而复合材料的吸附性能呈现减弱的趋势。因此,170 ℃为反应的最优碳化温度,所测得的吸附值为45.33 mg/g。

图8 碳化温度对复合材料吸附量的影响趋势曲线
3.3 响应面实验数据分析
根据单因素实验所得的数据作为响应面试验各项条件和因素设置的基础。并且控制催化剂用量、亚甲基蓝溶液浓度等变量,防止因素干扰[20]。以复合材料对亚甲基蓝的吸附量(y)为响应值,并选定单因素实验所考察的对响应值(复合材料对亚甲基蓝的吸附量)影响较大的3个因素(工艺)为响应面实验的变量,且设定葡萄糖和海泡石质量比为因素A(选定为1.0∶1.0、1.5∶1.0、2.0∶1.0),碳化时间为因素B(选定为4、8、12 h),碳化温度为因素C(选定为160、170、180 ℃)。最终,根据 Box-Benhnken的中心组合实验设计原理设计出了3因素3水平响应面试验,共17组实验,组成响应面实验因素水平设计表如表1所示,最终响应面实验设计以表2展示。
表1 响应面实验因素水平设计表
Table 1 Design table of factors and levels of response surface experiments

因素水平-101A1.0∶1.01.5∶1.02.0∶1.0B/h4812C/℃160170180
利用Design-Expert软件分析处理响应面实验所得到的数据,即得到响应面实验数据方差分析结果列于表3。由表3可见,P值和F值可以判断在实验中各变量对响应值(复合材料对亚甲基蓝的吸附量)影响的显著性。通过F检测进行影响显著性判断,在P值<0.01的情况下,判定变量对响应值的影响极显著,若P值<0.05,则认为该变量对响应值的影响显著,当P值>0.05,则变量对响应值影响不显著。
因此,由“Model(模型)”和“失拟项”可以看出,此次响应面实验模型的F值>0.05,P值<0.00001,失拟项大于0.05为不显著,说明模型相关度好可以用于吸附值预测。同时,发现A、B、C、AB、A2、B2、C2项对响应值的影响皆为极显著。
表2 响应面实验设计及相对应响应值表
Table 2 Response surface experimental design table and corresponding response values
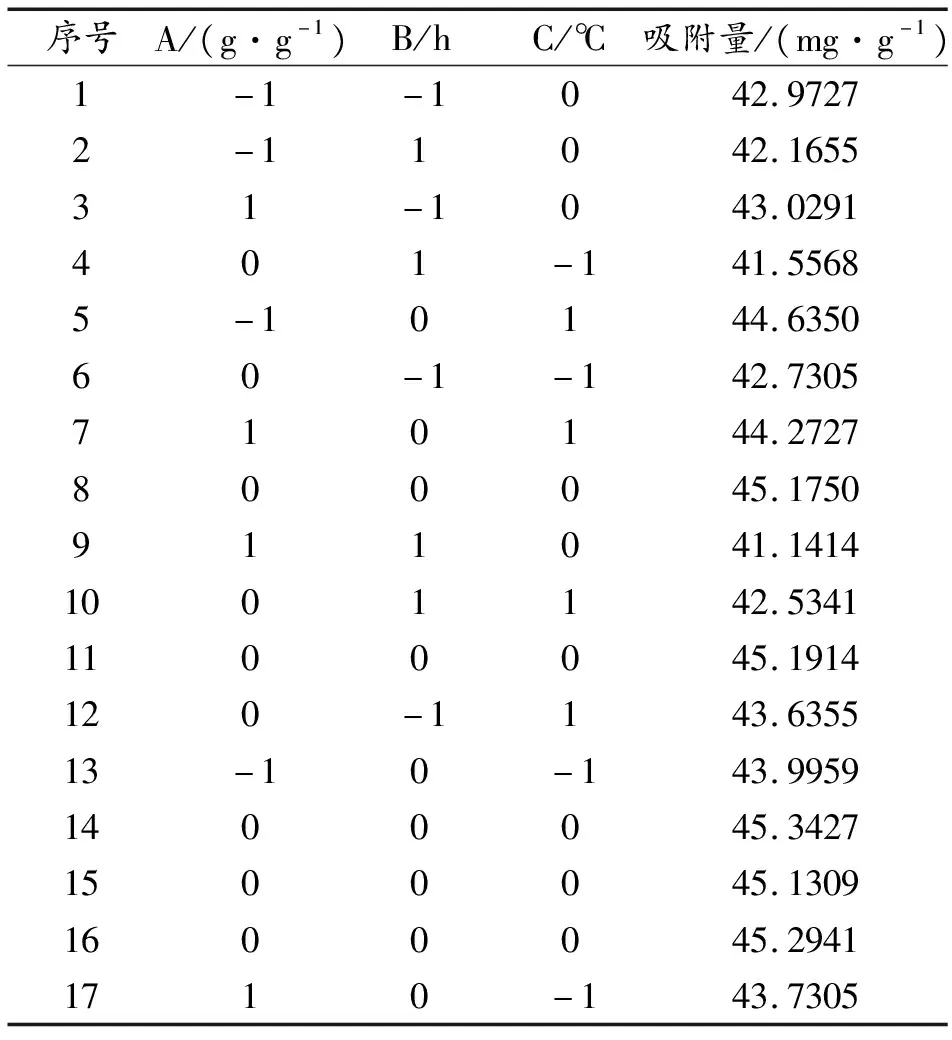
序号A/(g·g-1)B/hC/℃吸附量/(mg·g-1)1-1-1042.97272-11042.165531-1043.0291401-141.55685-10144.635060-1-142.7305710144.2727800045.1750911041.14141001142.53411100045.1914120-1143.635513-10-143.99591400045.34271500045.13091600045.29411710-143.7305
回归的响应面二次多项式为式(2):
Y=45.23-0.20*A-0.62*B+0.38*C-0.26*A*B-0.024*A*C+0.018*B*C-0.68*A2-2.23*B2-0.39*C2
(2)
响应面试验分析所得,复合材料对亚甲基蓝的吸附量实测值与预测值对比图,如图9所示。根据该图发现,各项实验数据基本上落在了中心线上,进一步说明,该响应面实验模型的拟合程度高,适合优化工艺条件的实验预测。
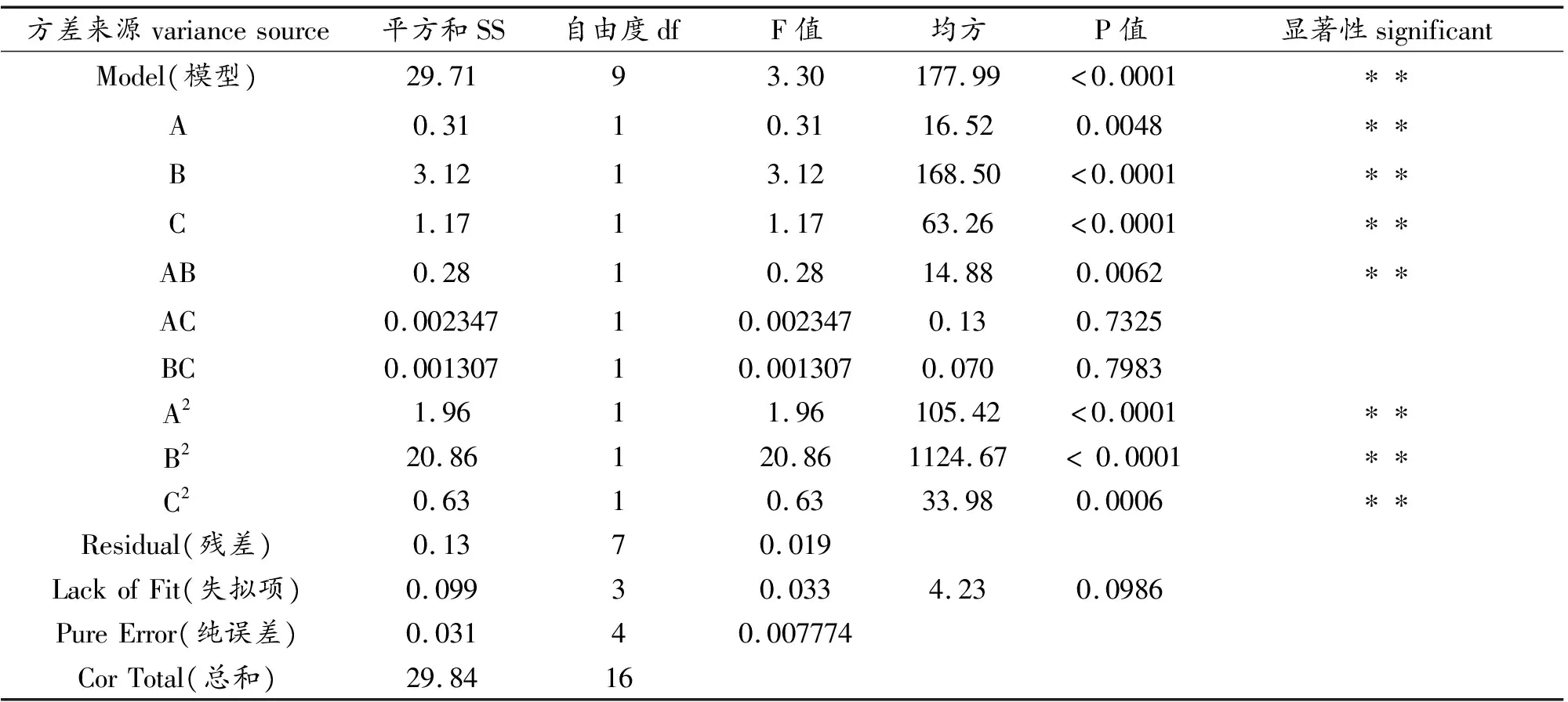
表3 响应面实验数据方差分析表
注:表中*代表显著(P值<0.05);**代表极显著(P值<0.01)。
图10~12展示的是3D响应曲面图与等值线图。由图可见,在实验范围内,葡萄糖与海泡石的质量比(A)、碳化时间(B)及碳化温度(C),都出现出了大致相同的变化趋势,当因素水平比较低时,复合样品的吸附量水平较低,而随着因素水平的提高,吸附量呈现出了先向高水平上移之后再下降的趋势,该结果与单因素实验相似。其中,AB的交互作用最为显著。
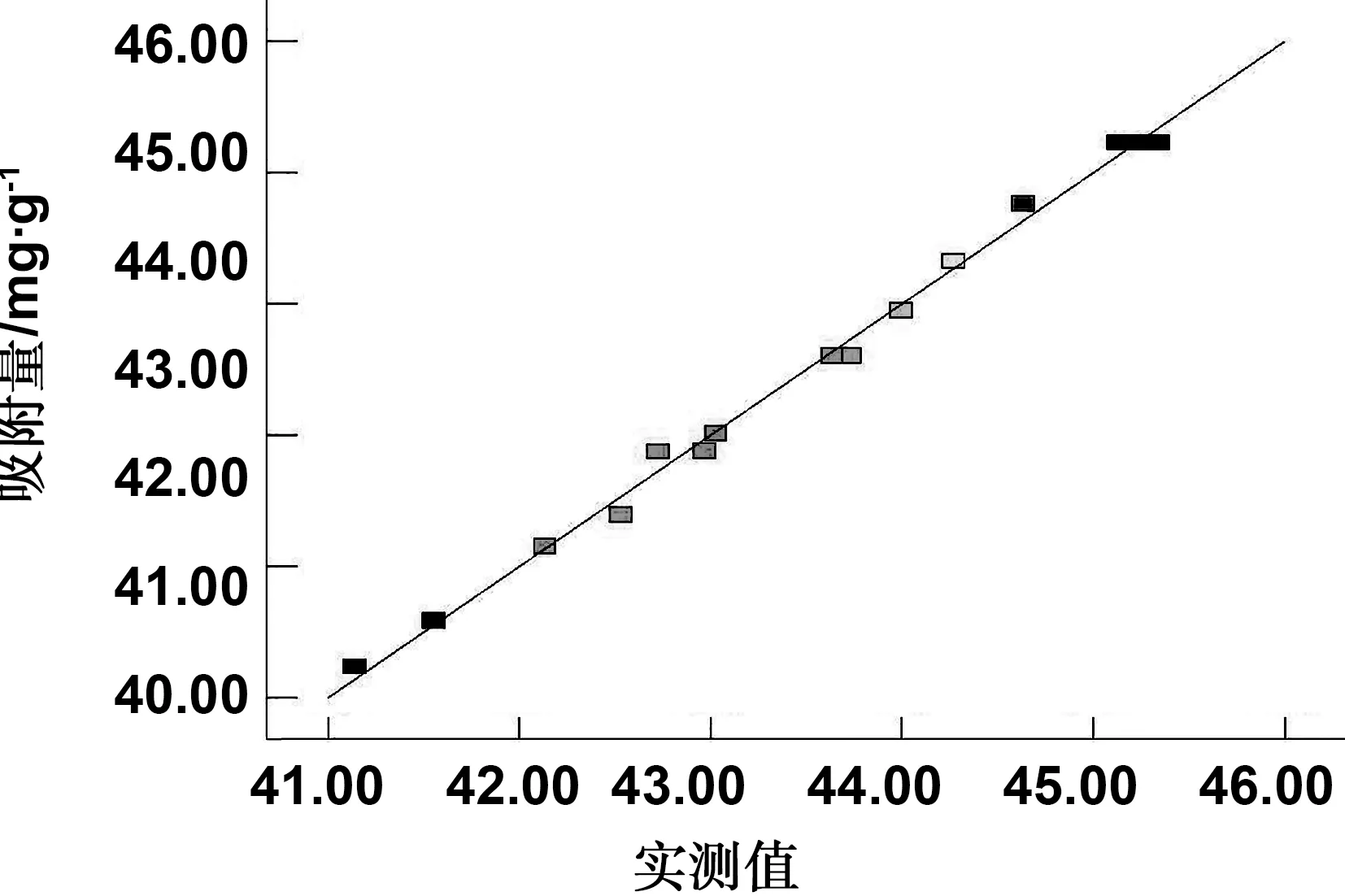
图9 复合材料对亚甲基蓝的吸附量实测值与预测值对比
通过对图10的3D响应曲面图与等值线图分析,发现同时碳化时间对复合材料吸附亚甲基蓝量的影响大于葡萄糖与海泡石的质量比的影响;对图11进行分析发现碳化温度对复合材料吸附亚甲基蓝量的影响大于葡萄糖与海泡石的质量比的影响;经由图12,则可发现碳化时间对复合材料吸附亚甲基蓝量的影响大于碳化温度的影响。
因此,结合响应面实验数据方差分析结果中的F值和P值、以及各因素间3D响应曲面图与等值线图,发现葡萄糖和海泡石质量比、碳化时间、碳化温度三个因素对亚甲基蓝的吸附量的影响大小依次为碳化时间 > 碳化温度 > 葡萄糖与海泡石质量比,各因素对响应值的影响不是简单的线性关系。
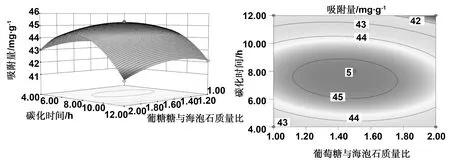
图10 质量比与碳化时间(AB)间交互作用的3D响应曲面图与等值线图

图11 质量比与碳化温度(AC)间交互作用的3D响应曲面图与等值线图

图12 碳化时间与碳化温度(BC)间交互作用的3D响应曲面图与等值线图
经由响应面实验结果所得葡萄糖碳化物/海泡石复合材料工艺最优工艺条件为:葡萄糖与海泡石质量比为1.49∶1.0;碳化时间为7.37 h;碳化温度为175.21 ℃,预测最优吸附量为45.367 mg/g。
因此,修改葡萄糖碳化物/海泡石复合材料制备工艺条件为葡萄糖与海泡石质量比为1.49∶1.0、碳化时间为7.33 h(即440 min)、碳化温度为175 ℃,进行验证实验,将结果列于表4中。

表4 验证实验结果
实测吸附量稍低于预测值0.32 %。因此,对此次响应面试验进行总结,认为该数学模型能够较为准确地预测葡萄糖碳化物/海泡石复合材料对亚甲基蓝吸附量的预测,可用于复合材料的工艺优化实验。
4 结 论
(1)本实验成功制备出葡萄糖碳化物/海泡石复合材料,扫描电镜发现有均匀的碳化微球非均匀分布在海泡石纤维的表面,直径约为0.10~0.15 μm;红外光谱图上表明有饱和羟基的 C—H、C=O 和 C=C双键的存在;XRD图谱表明材料复合前后,晶型不发生改变;BET分析结果,符合随着负载量的增加而比表面积逐渐变小的趋势。因此,综合说明,葡萄糖碳化物成功负载于海泡石表面。
(2)采用响应面法确定葡萄糖碳化物/海泡石复合材料工艺最优工艺条件为:葡萄糖与海泡石质量比为1.49∶1.0、碳化时间为7.33 h(440 min);碳化温度为175 ℃,最优吸附量为 45.22 mg/g,吸附效果优于经过相同工艺条件水热碳化处理的海泡石原材料(吸附量为35.13 mg/g)。
(3)在负载了葡萄糖碳化物之后,复合材料的比表面积明显小于海泡石的比表面积,但复合材料对亚甲基蓝的吸附能力明显高于海泡石,说明了葡萄糖碳化物负载在海泡石表面后,所引入的C—H、C=O 和C=C等有机官能团,增强了复合材料的吸附能力。
猜你喜欢
上海金属(2022年6期)2022-11-25
热处理技术与装备(2022年5期)2022-10-26
四川水泥(2022年9期)2022-09-24
环境卫生工程(2021年4期)2021-10-13
环球时报(2020-10-19)2020-10-19
合成树脂及塑料(2020年3期)2020-01-16
模具制造(2019年3期)2019-06-06
重庆建筑(2019年12期)2019-02-18
陶瓷学报(2018年4期)2018-09-13
资源环境与工程(2014年5期)2014-01-16
