
碱金属离子在蒙脱石-Cu2+表面吸附的离子特异性*
2020-04-25 01:47潘小丽刘新敏
土壤学报 2020年2期
潘小丽,刘新敏,李 航,李 睿
碱金属离子在蒙脱石-Cu2+表面吸附的离子特异性*
潘小丽,刘新敏,李 航,李 睿†
(西南大学资源环境学院,土壤多尺度界面过程与调控重庆市重点实验室,重庆 400715)
研究离子交换中的离子特异性效应有助于揭示离子-带电表面相互作用机制。以蒙脱石Cu2+饱和样为研究对象,采用恒流法研究不同浓度碱金属离子Li+、Na+和K+的吸附动力学过程,并建立1︰1型(LiNO3、NaNO3、KNO3)电解质溶液中离子平衡吸附量与体系吸附活化能之间的关系。结果发现:(1)Li+、Na+和K+在蒙脱石-Cu2+表面的吸附过程仅呈现出弱静电力作用下的一级动力学特征,并存在明显的离子特异性效应。(2)离子非经典极化作用与体积效应共同决定了离子在双电层中的位置,从而导致表面电位存在差异;并且表面电位(绝对值)随着电解质浓度降低而增加,表现为Li+> Na+> K+。(3)根据新建立的模型可预测吸附离子在双电层中的位置,进而求出体系的吸附活化能,并发现离子特异性效应产生的根本原因是由活化能决定的,同时本研究表明建立的新模型在固/液界面反应中具有普适性。本研究将对固/液界面反应理论的完善提供新思路。
带电表面;离子吸附;表面电位;非经典极化;活化能;离子特异性
离子特异性效应最早发现于不同盐溶液对蛋白的聚沉实验[1],指同价离子在反应体系中表现出不同的特征或对系统产生不同的影响[2],离子特异性效应普遍并深刻影响着一系列物理、化学与生物过程[3-5]。2012年Lo Nostro和Ninham[6]在中指出,离子特异性效应下蕴藏的科学基础将是物理化学这座大厦坚实的基础。
在矿物颗粒/水界面反应研究中,Liu等[7]发现Ca2+/Na+在伊利石表面的交换过程表现出强烈的离子特异性效应,并且该过程受到表面电场的影响。Du等[8-9]研究了不同碱金属离子在蒙脱石表面的吸附动力学,也发现吸附动力学过程存在强烈的离子特异性效应,并用离子非经典极化理论做出了解释。土壤/黏土矿物颗粒表面带有的大量电荷使颗粒表面形成数量级达108~109V·m–1的强电场[7,10],离子受强电场影响,其外层电子能量和量子状态发生根本性改变,因此产生强烈的非经典极化效应[7]。由于极化效应而产生的额外势能(Hofmeister能量)导致了离子在交换/吸附过程中呈现出强烈的离子特异性效应[7,11]。Li+、Na+和K+均为第一主族元素,在矿物颗粒表面吸附过程中不发生价态变化和溶解度较高。并且,相比其他价态离子,碱金属离子在矿物颗粒表面多为静电吸附,易于阐明离子界面反应中的吸附机理。本研究的实验材料选取蒙脱石是因为:(1)该矿物边缘处的电荷占表面总电荷百分比非常小,因此在给定条件下,其表面电荷可视为常数;(2)蒙脱石属于恒电荷表面,碱金属离子与表面边缘处的化学键作用相比于静电吸附作用可以忽略;(3)蒙脱石矿物的表面相对光滑,因此表面能态非常简单;(4)其矿物结构在实验中更稳定。
目前大量研究均集中于碱金属/碱土金属离子的吸附动力学研究[8-9,12],尚未涉及离子特异性对重金属离子吸附/解吸的影响。然而,重金属离子在土壤中积累、传输、扩散却是导致环境问题的一大原因。当重金属离子被黏土矿物吸附至其表面,并与溶液达到平衡后,一旦溶液中的离子组成或浓度发生改变,吸附于黏土矿物的重金属离子也有可能被其他离子交换而进入土壤溶液中,造成环境的二次污染[13]。因此本研究选取Cu2+为代表,以Cu2+表面饱和的蒙脱石为材料,对几种碱金属离子(Li+、Na+、K+)在蒙脱石-Cu2+表面的吸附动力学进行分析,阐明碱金属离子在蒙脱石-Cu2+表面的吸附机制,为进一步明确离子-带电表面相互作用机制提供理论基础。
1 材料与方法
1.1 实验材料与样品的制备
硝酸铜、硝酸钾、硝酸钠和硝酸锂均为分析纯(成都市科龙化工试剂厂);本实验中采用蒙脱石(内蒙古物华天宝矿物资源有限责任公司)作为研究材料。用0.1 mol·L–1的Cu(NO3)2溶液对蒙脱石样品进行表面处理,制成Cu2+饱和样,制作方法见参考文献[12]。通过联合测定法[10]测得蒙脱石表面电荷数量为115 cmol·kg–1,即本实验Nmax)=115 cmol·kg–1。比表面积为725 000 m2·kg–1,表面电荷密度为0.15 C·m–2。
1.2 实验方法
采用恒流法进行离子交换/吸附实验[14]。在298 K的恒温条件下,0.000 1、0.001、0.01 mol·L–1的KNO3、NaNO3、LiNO3以及0.1 mol·L–1的KNO3交换液均以恒定流速(1 mL·min–1)流过平铺的实验样品,用自动部分收集器(DBS-100,上海沪西分析仪器厂)按一定设定时间间隔收集流出液,由于交换液浓度增大后离子吸附速率加快,离子交换时间缩短,因此交换液浓度为0.000 1、0.001、0.01、0.1 mol·L–1时对应时间间隔分别为10、5、3、2 min。用火焰光度计(AP1401,上海傲谱分析仪器有限公司)分别测定流出液中 K+、Na+、Li+的浓度,从而计算出不同时间间隔的K+、Na+、Li+的吸附量;用原子吸收分光光度计(Z-5000型,日立,日本)测定Cu2+的浓度,从而计算出不同时间间隔Cu2+的解吸量。
1.3 数据处理
X+/Cu2+(X+代表Li+、Na+、K+)交换体系中,当离子X+吸附达到平衡时,阳离子在扩散层中的分布可通过图1描述:
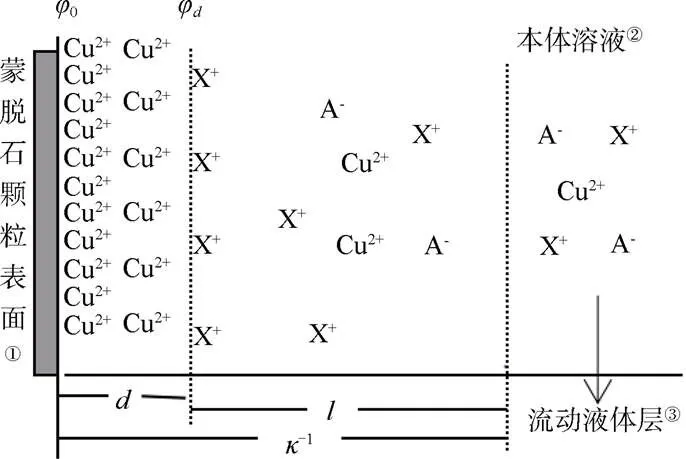
注:d为吸附平衡时离子与黏土矿物表面间的距离;l为扩散距离;κ–1为双电层的厚度;φ0为表面电位;φ d为离子距表面d处的电位;A–为阴离子。 Note:d stands for distance of ions from the surface of clay minerals at adsorption equilibrium;l for diffusion distance;κ–1 for thickness of the double electric layer;φ0 for surface potential;φd for the potential where ions d away from the surface;and A– for anion. ① Montmorillonite particle surface,② Bulk solution,③ Flow solution layer
Li等[15-16]建立了考虑离子特异性效应的离子吸附动力学模型,当土壤颗粒表面对离子吸附存在快速的强吸附作用时,可能存在零级动力学过程。然而,当表面由于强静电力吸附作用吸附至饱和后,离子将在弱静电力作用下继续扩散,动力学吸附过程表现为一级动力学特征。离子在蒙脱石表面的平衡吸附量可表达为[17]:
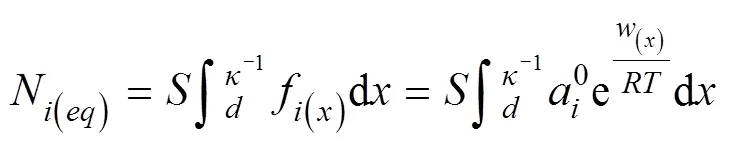
式中,N()为由实验结果得到的平衡吸附量,mmol·g–1;为比表面积,m2·g–1;为Debye常数;为碱金属离子与黏土矿物表面间的距离,dm;f(x)为离子在扩散层中处的浓度,mol·L–1;a0为离子在本体溶液中的活度,mol·L–1;为气体常数;为绝对温度,K;(x)为位置处离子的吸附能,kJ·mol–1。当离子达到平衡时,考虑离子间相互作用的Boltzmann分布函数可表达为[18]:
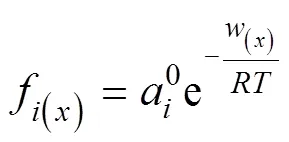
当考虑Hofemister附加能量时[11]:
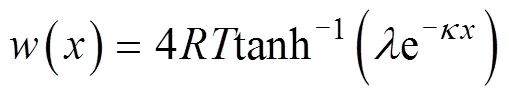
其中,
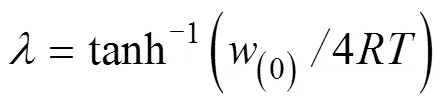

w(0)为阳离子在颗粒表面的Hofemister能量,kJ·mol–1;Z为离子的化合价;为法拉第常数,C·mol–1。将式(2)代入式(1)中,得:
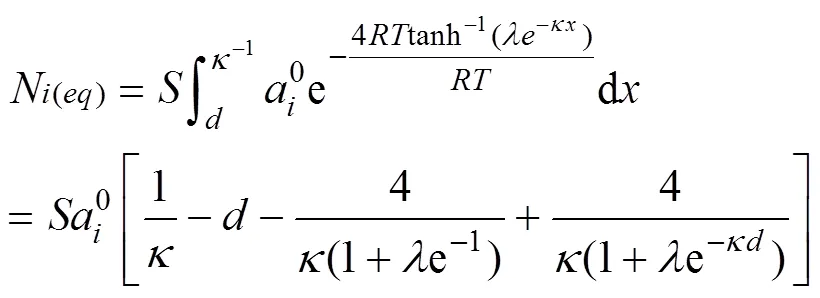
当=0时,表明离子扩散至蒙脱石颗粒表面,此时吸附量达到最大值N(max),N(max)代表因同晶替换所产生的负电荷全部被碱金属离子中和时该碱金属离子的潜在最大吸附量,mmol·g–1。

由式(7)求解得到,进而由式(6)计算出各实验条件下离子到达表面的位置,因此通过式(8)即可求得各实验条件下的吸附活化能,kJ·mol–1。

由式(5)可知,(0)随表面电位增大而增大,当(0)→–∞时,由式(4)可知→–1,体系处于高电位(绝对值)条件下,2Sa/κN(eq)1,此时联立式(6)、式(7)和式(8)得到:

式中,N(eq)/N(max)表示离子在蒙脱石表面的吸附饱和度。式(9)即为式(8)在高电位(绝对值)条件下的离子吸附饱和度与活化能之间的理论关系,结合化学反应中Arrhenius公式[19],发现二者在形式上一致,说明本研究中提出的离子吸附模型具有普适性。
2 结果与讨论
2.1 不同浓度碱金属离子在蒙脱石-Cu2+表面的吸附量
不同碱金属离子Li+、Na+、K+在蒙脱石表面吸附量随时间的变化如图2所示。由图2可知,尽管Li+、Na+和K+是同价离子,但是在相同电解质浓度条件下,相同时间段内Li+、Na+和K+的累积吸附量并不相同:(1)当电解质浓度相等时,相同时间内K+的累积吸附量明显高于Li+和Na+,呈现出离子特异性效应,且累积吸附量随时间的增加而增加;(2)相同时间段内,不同浓度条件下不同离子的吸附量亦不同,且累积吸附量随电解质浓度的增加而增加。例如,当离子浓度为0.000 1 mol·L–1、吸附时间为20 min时,K+、Na+和 Li+的吸附量分别为2.89、2.34和1.31 mmol·kg–1;当离子浓度为0.001mol·L–1、吸附时间为20 min时,K+、Na+和 Li+的吸附量分别为26.0、19.2和19.1 mmol·kg–1。当离子浓度为0.01 mol·L–1、吸附时间为18 min时,K+、Na+和 Li+的吸附量分别为254、105和138 mmol·kg–1。
为了直接利用实验数据,将动力学方程写成差分形式,并直接利用动力学实验数据可得到dN()/d. N()间的关系,结果如图3所示。同时根据图3所获得的吸附动力学参数结果列于表1中,由图3和表1可以看出:(1)不同浓度条件下的碱金属离子K+、Na+和Li+在交换Cu2+时均呈现出一级动力学特征,说明该条件下碱金属离子K+、Na+和Li+在蒙脱石-Cu2+表面下的吸附为弱静电力作用下的吸附。Du等[9]提出强极化离子的吸附将在吸附初期表现出零级速率特征,而后变为一级速率特征,但是弱极化离子仅表现出一级速率特征,并且吸附动力学过程中存在的离子特异性效应用Hofmeister能H(0)来定量描述,离子的H(0)较高时,在吸附初期会出现零级动力学过程,随后再转化为一级动力学过程;反之,H(0)较低时,吸附仅出现一级动力学过程。由此可推测本实验体系中K+、Na+和Li+的 Hofmeister能H(0)相比Cu2+的均较低。(2)相同电解质浓度条件下K+、Na+和Li+的吸附动力学速率和平衡吸附量均存在离子特异性效应,并且电解质浓度影响离子的吸附选择性顺序,例如,电解质浓度为0.000 1 mol·L–1与0.001 mol·L–1时平衡吸附量表现为K+>> Na+> Li+;当浓度升高至0.01 mol·L–1时,平衡吸附量则呈现为K+>> Li+> Na+,表明在较高浓度下离子的体积效应可能对平衡吸附量存在影响。
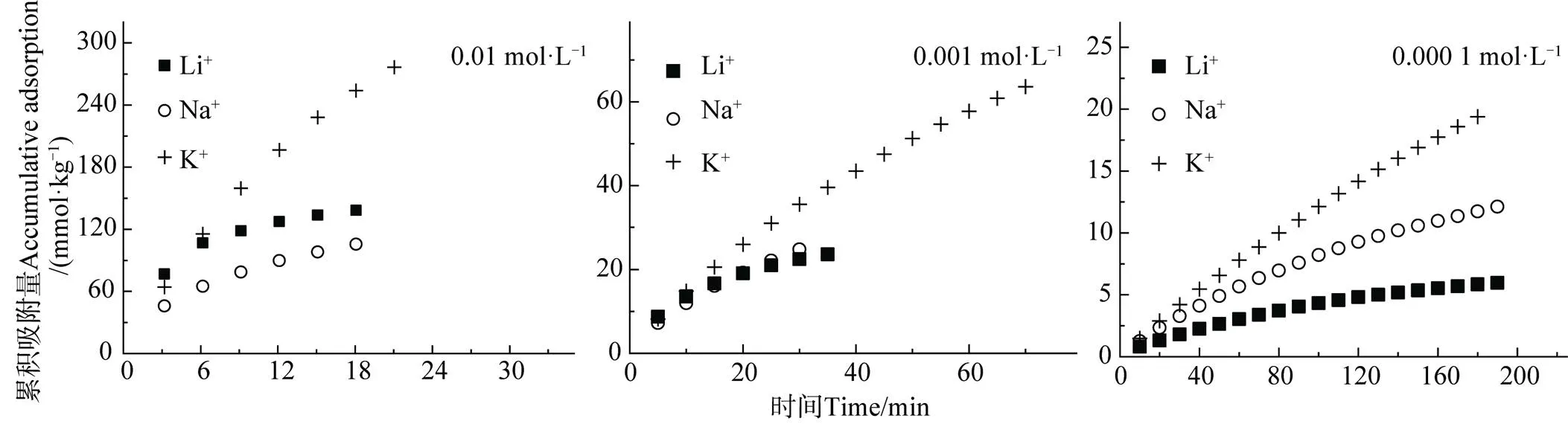
图2 不同浓度碱金属离子累积吸附量(Ni)随时间的变化
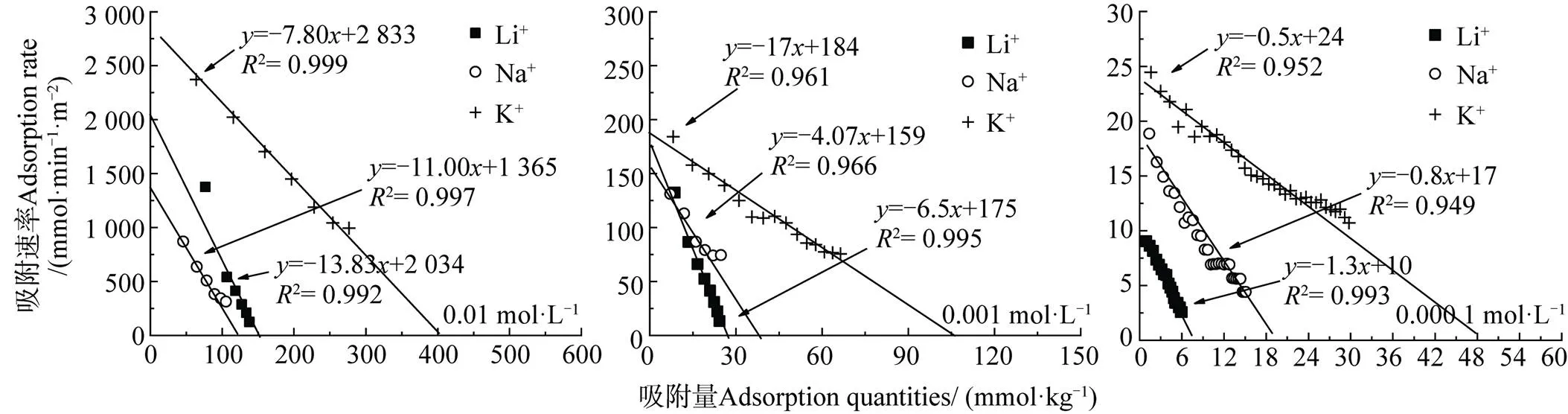
图3 不同浓度碱金属离子在蒙脱石颗粒表面的吸附动力学关系曲线
表1中还列出了0.000 1 mol·L–1、0.001 mol·L–1、0.01 mol·L–1浓度条件下Li+、Na+交换蒙脱石-K+饱和样的相关动力学数据,从表1中可以看出,即使在相同浓度以及相同类型电解质条件下,Li+、Na+在蒙脱石Cu2+饱和样表面与在K+饱和样表面的吸附速率与平衡吸附量均存在明显差异:尽管相同浓度条件下的Li+、Na+离子在两种饱和样表面的离子吸附量与吸附速率均随电解质浓度的增加而增加,但是Li+、Na+离子在蒙脱石-K+饱和样表面的平衡吸附量是相应浓度条件下Cu2+饱和样表面平衡吸附量的2倍~10倍,并且平衡吸附量差异随着电解质浓度的降低而逐渐增大。表明相同离子类型和电解质浓度条件下,矿物表面被饱和的离子类型会强烈影响其他离子在表面的吸附能力。相应地,碱金属离子的吸附必定会引起蒙脱石表面Cu2+的解吸,图4a)为电解质浓度在0.001 mol·L–1条件下K+、Na+和Li+交换蒙脱石-Cu2+饱和样,利用解吸动力学理论[20]得到的Cu2+解吸动力学结果。解吸过程仅表现出一级动力学过程,并且该过程也呈现出明显的离子特异性效应,且碱金属离子的吸附量越大对应的Cu2+解吸量也越大,平衡解吸量与吸附量均表现出K+>> Na+> Li+的趋势。但是本研究发现,一价离子的吸附量不等于Cu2+的解吸量乘以ZCu(ZCu=2),可能是由于被解吸的Cu2+在溶液中出现了羟基酮。蒙脱石属于恒电荷表面,其矿物边缘处的可变电荷占表面总电荷百分比非常小[21],因此重金属离子在蒙脱石表面发生的专性吸附可忽略不计。图4b)是0.1 mol·L–1KNO3交换蒙脱石-Cu2+的实验结果,发现此浓度下Cu2+解吸量达到了572 mmol·kg–1,基本达到了蒙脱石表面的电荷数量,说明Cu2+在蒙脱石表面的专性吸附量非常小,因此本文中的蒙脱石表面电荷数值可用最大潜在吸附量N(max)值代替。
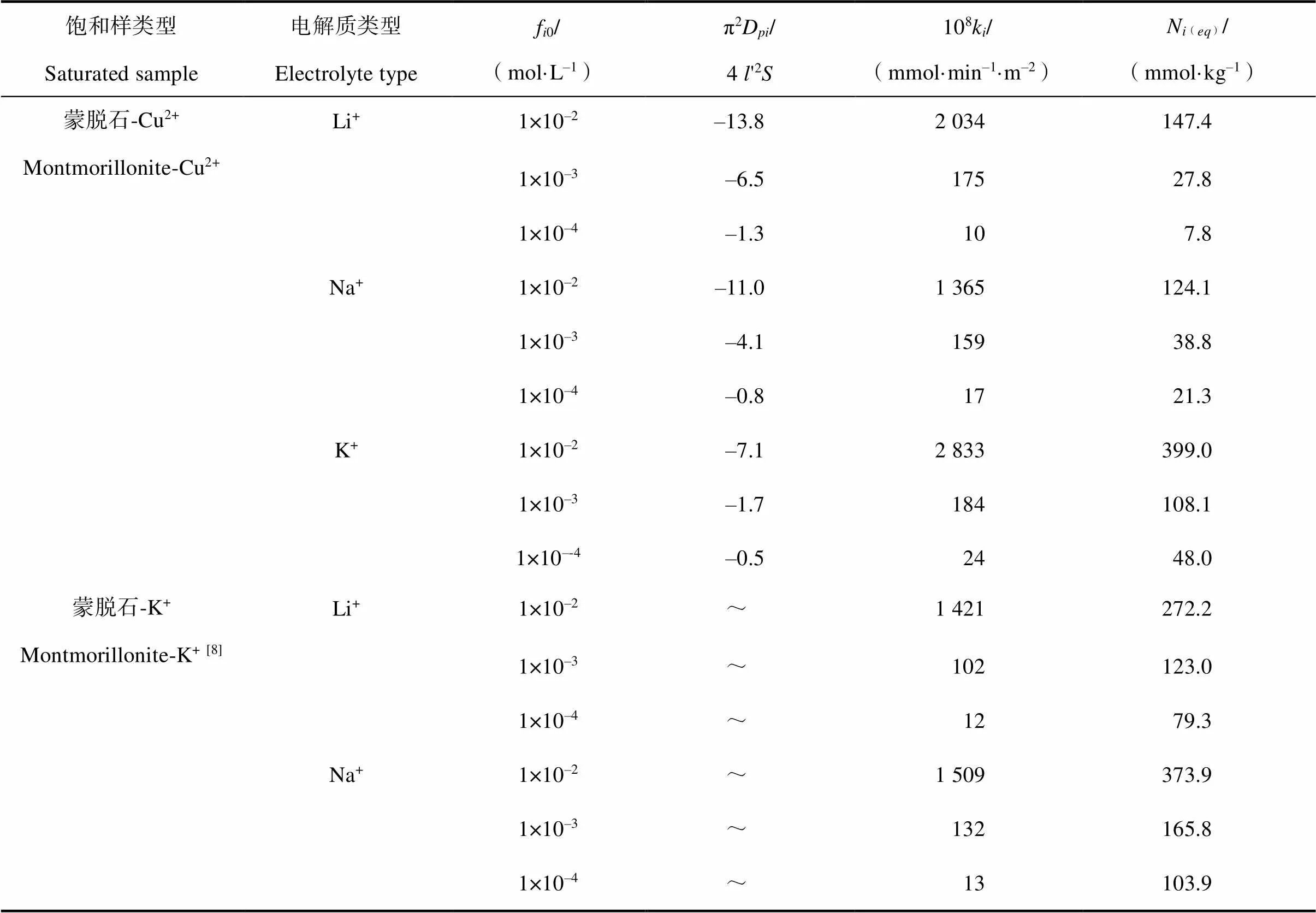
表1 离子吸附速率方程中相关参数
注:f0表示电解质浓度;π2D(4'2)–1表示速率方程的斜率;k表示离子动力学吸附速率;N(eq)表示离子平衡吸附量。下同 Note:f0stands for electrolyte concentration;π2D(4'2)–1forslopeof the rate equation;kfor kinetic adsorption rate;and N(eq)for equilibrium adsorption quantities. The same below
刘汉燚等[22]运用原子力显微镜观察测定几种碱金属离子氯化盐溶液处理的蒙脱石颗粒在风干条件下的形貌图和黏附力时也发现,不同类型电解质条件下蒙脱石颗粒的凝聚也存在明显的离子特异性效应,并指出影响颗粒聚集中的离子特异性效应的主要因素为离子体积和量子涨落效应;Tian等[23]在研究蒙脱石矿物的相互作用中也发现了离子特异性并采用活化能解释了离子特异性效应。本实验在不同电解质浓度条件下也出现了离子特异性效应,但是仅用高浓度下起作用的离子大小、水合作用和色散力并不能完全、合理地解释实验结果[24]。而在固/液界面中,另一个至关重要的作用力,即离子的非经典极化作用(量子涨落效应),也强烈影响着离子与表面的相互作用[25],进而影响着离子的吸附动力学行为。由式(5)~式(9)可以看出,平衡吸附量与表面电位以及活化能有关,因此接下来进一步计算不同电解质浓度及电解质类型条件下的表面电化学参数以及活化能来探究其原因。
2.2 不同电解质浓度下碱金属离子吸附过程中表面电位及活化能
Li等[26]利用激光闪射实验研究蒙脱石胶体活化能,估算了Na+在颗粒表面的相对有效电荷系数Na(0)=1.18,Liu等[11]通过研究电解质浓度范围在0.001~0.03 mol·L–1的Na+/K+和Na+/Li+交换实验发现,相对有效电荷Li(0)/Na(0)=0.901,K(0)/Na(0)=1.646。因此,本研究分别得到Li(0)=0.901×1.18=1.063,K(0)=1.18×1.646=1.942。根据式(7)可求出各个体系的,进而代入式(6)可计算吸附平衡时不同体系中碱金属离子与黏土矿物表面间的距离(图5),最后可获得交换吸附平衡时吸附离子实际扩散距离,=–1–。
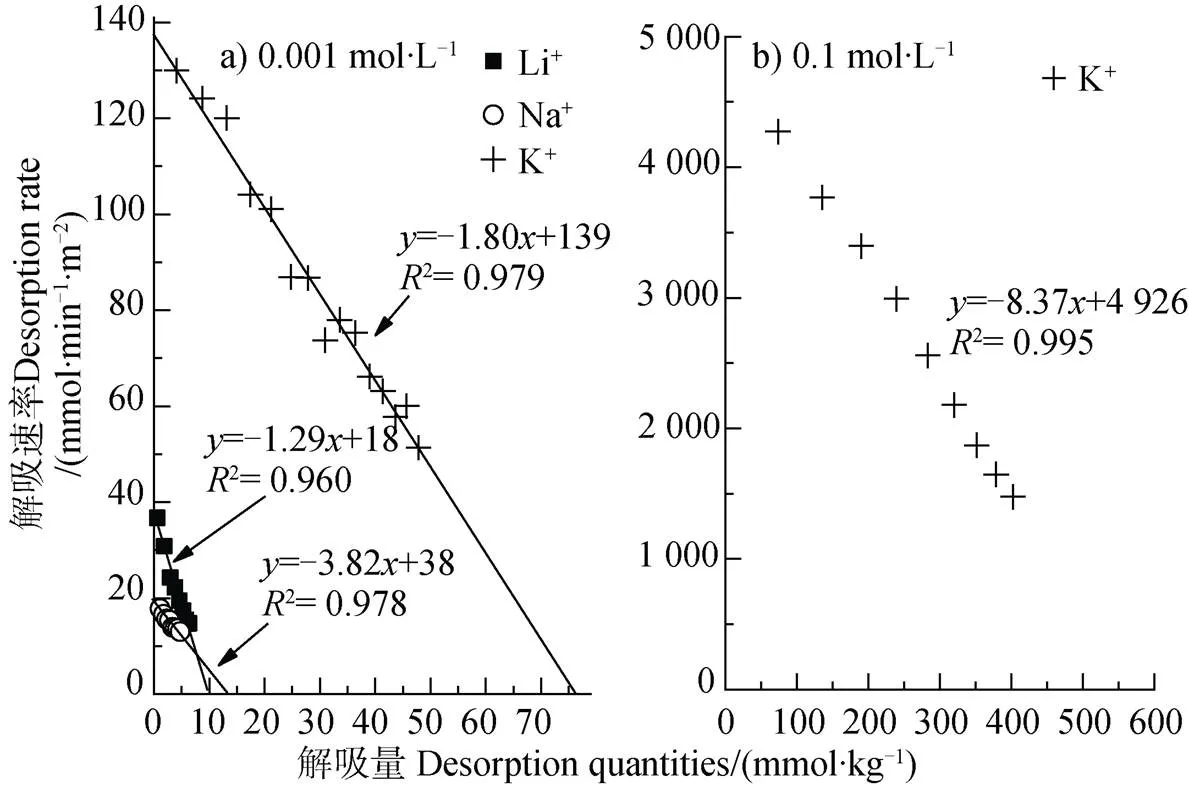
图4 碱金属离子在蒙脱石颗粒表面的解吸动力学关系曲线
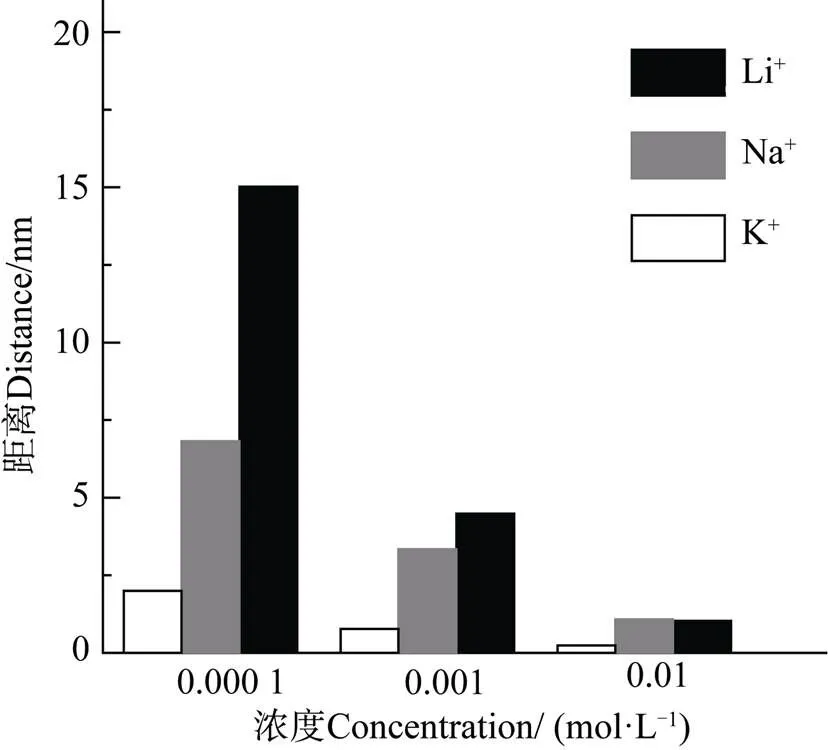
图5 不同浓度碱金属离子电解质溶液中离子距离黏土矿物表面的距离
由图5可知:(1)在同一电解质体系中,随着电解质浓度的增加,减小,说明离子距离蒙脱石颗粒表面更近;这是因为体系的吸附能随离子浓度增大而增加,使得吸附过程中需克服的势垒逐渐降低,离子能够到达离矿物表面更近的地方,这与Brown等[27]研究结果吻合。吸附平衡时离子实际扩散距离越大,越有利于离子与蒙脱石表面的Cu2+进行交换,从而导致吸附量增加。(2)在电解质浓度为0.000 1 mol·L−1和0.001 mol·L–1的体系中,均有Li>Na>K;当浓度升高至0.01 mol·L–1时,Li+相比Na+能够到达离表面更近的位置,表现为Na>Li>K。这是由于K+外层电子层数最多,在颗粒表面的非经典极化作用显著强于Na+和Li+,非经典极化作用使K+更容易趋于靠近蒙脱石颗粒表面,与表面吸附的Cu2+实现更多的交换,导致K+吸附量远大于Na+和Li+。另一方面,根据Stern层理论,体积效应也显著影响双电层的结构。随着电解质浓度升高,体积效应逐渐增强,尽管Na+极化作用较Li+强,但相比K+其极化作用较弱,Na+不能克服高浓度条件下自身较强的体积效应,从而导致Na>Li,Li+能与表面吸附的Cu2+实现更多的交换,从而解释了图3中0.01 mol·L–1浓度条件下离子吸附量大小序列与其他两个浓度不同。(3)随着浓度减小,三种离子间值差异逐渐增大。这是因为电解质浓度越低,离子对表面电场的屏蔽能力越弱,表面电位(绝对值)增大,表面与吸附离子之间的非经典极化作用增强,最终导致三种离子间值差异变大。说明低浓度下,非经典极化对离子扩散距离的影响占主导作用。随着电解质浓度增加,离子对表面电场的屏蔽能力增强,离子的非经典极化作用逐渐减弱,离子间体积效应逐渐增强,导致三种离子间值差异逐渐缩小。因此,非经典极化与体积效应共同决定了离子在固/液界面的扩散距离。
已知()=γZFφ,其中γ为离子的相对有效电荷系数,结合式(3)得到:
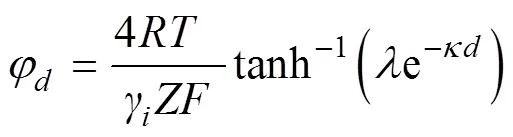

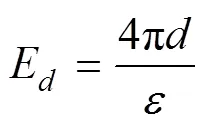
式中,为距离矿物表面处的电荷密度,C·m–2;E为距离矿物表面处的电场强度,mV·m–1;为水的介电常数,8.9×10–10C2·J–1·dm–1。当0时,即为颗粒的表面电位(0):

将不同交换体系中的表面电化学参数计算结果列于表2。由表2得出:(1)同一电解质类型,颗粒的表面电位0(绝对值)随着体系电解质浓度的降低而升高,也说明低浓度电解质对蒙脱石颗粒表面电场的屏蔽作用较弱。0随体系电解质浓度变化幅度大于,因为距离表面越近,电位梯度越大。(2)各体系中值均小于表面电位0,0约为的1倍~5倍,且两者的差异随着电解质浓度降低而增大。(3)离子吸附平衡时离子距离表面位置处的电荷密度和电场强度E随着电解质浓度的升高而增大,是因为离子随电解质浓度升高后,扩散距离增大,导致其电荷密度及电场强度增大。电场强度E高达107~108V·m–1的数量级,说明固/液界面交换体系的确处在颗粒表面强电场中,该电场强烈影响离子交换过程。
从表1中还可以发现:(1)在不同电解质浓度条件下,K+、Na+和Li+在蒙脱石-Cu2+表面的吸附饱和度不同,并未达到100%;(2)阳离子类型和浓度显著影响吸附饱和度,即离子在蒙脱石-Cu2+表面的吸附饱和存在明显的离子特异性效应;(3)即使在静态恒流实验中,真正的平衡也很难实现,特别是对于离子体积较小的Na+和Li+。

表2 吸附实验中各交换体系的表面电化学参数
注:0为表面电位;φ、、分别为吸附平衡时离子实际到达处位置的电位、电荷密度和电场强度。Note:0stands for surface potential;φ,,andfor potential,charge density and electric field intensity,respectively,in the positions the ions actually arrive at when adsorption reaches equilibrium
根据式(9)与式(8)可分别求出活化能理论预测值与实验值,并用离子吸附饱和度与活化能作图,结果如图6所示:(1)当活化能为零时,离子吸附饱和度接近于100%,说明离子扩散至蒙脱石颗粒表面,此时交换完全,吸附量N(eq)达到最大值N(max),实验求得的活化能符合理论预测值,进一步验证了本模型理论的正确性。(2)三种离子间活化能存在明显的离子特异性效应,K+体系中最低,因此离子扩散距离最远,与蒙脱石表面吸附的Cu2+交换更彻底,对应的吸附量值最大。(3)Li+、Na+和K+吸附饱和度均随着活化能的降低而增大,这是由于Li+、Na+和K+的吸附能随离子浓度的增大而增加,当Li+、Na+和K+离子浓度增加后,体系中碱金属离子的吸附能增加,此时与蒙脱石颗粒表面Cu2+之间的能量势垒差减小,离子交换过程中的活化能减小,碱金属离子能与更多的Cu2+实现交换,大量进入扩散双电层的碱金属离子将蒙脱石表面吸附的Cu2+解吸下来。(4)尽管Li+、Na+和K+体系离子吸附饱和度存在特异性效应,但是不同离子间吸附饱和度与活化能均符合同一规律。表明碱金属离子在蒙脱石-Cu2+表面吸附的离子特异性效应本质上是由活化能决定的,并且通过本研究建立的离子吸附模型预测的活化能与化学反应中阿累尼乌斯定律所预测结果一致。
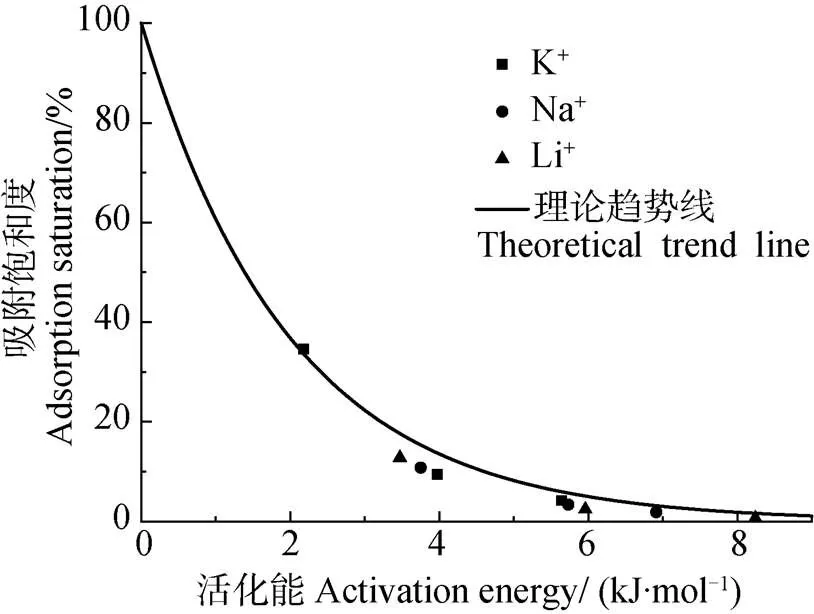
图6 不同体系中吸附饱和度与活化能关系
本研究选用的实验体系较单一,而自然土壤是多组分多离子的复杂体系,吸附机理更加复杂,今后应逐渐过渡到探讨多组分体系下的混合离子吸附机理,最后结合农业生产实际的土壤,开展原位模拟离子交换吸附研究。此外,土壤重金属污染治理是目前我国土壤污染治理的重点,黏土矿物、单硅酸盐及硅肥等硅酸盐钝化修复材料是重要的土壤重金属化学稳定剂[28],今后可结合农业生产实际,开发有利于农作物生长的长效硅酸盐土壤重金属钝化剂。离子和带电粒子之间的相互作用受纳米/胶体颗粒表面上离子吸附控制,同时它决定着颗粒的微观性质、界面反应过程以及颗粒间相互作用。本研究从机理上分析了固/液界面反应过程,有助于揭示离子-带电表面相互作用机制,进而为阐明土壤中的宏观现象,如土壤侵蚀、土壤中营养元素的迁移、农业污染等提供理论基础。
3 结 论
通过恒流法研究碱金属离子Li+、Na+和K+在不同浓度下交换蒙脱石-Cu2+的吸附动力学行为,并建立了1︰1型(LiNO3、NaNO3、KNO3)电解质溶液中,离子平衡吸附量与体系吸附活化能之间的关系。得到如下重要结论:碱金属离子K+、Na+和Li+在0.01 mol·L–1、0.001 mol·L–1和0.000 1 mol·L–1浓度下交换蒙脱石表面吸附的Cu2+时,吸附过程均仅呈现出弱静电力作用下的一级动力学特征并存在着明显的离子特异性效应,离子特异性效应可通过吸附速率与平衡吸附量来表征。碱金属离子的平衡吸附量与吸附速率均随着碱金属浓度的降低而降低。蒙脱石对碱金属离子的吸附存在强的选择性,且吸附选择性顺序受碱金属浓度的影响。相同离子类型和电解质浓度条件下,矿物表面被饱和的离子类型会强烈影响其他离子的吸附过程。非经典极化与体积效应共同决定了离子在固/液界面的扩散距离,低浓度下,非经典极化对离子扩散距离的影响占主导作用;随着电解质浓度增加,离子对表面电场的屏蔽能力增强,离子的非经典极化作用逐渐减弱,离子间体积效应逐渐增强,导致三种离子间扩散距离差异逐渐缩小。离子扩散距离的差异导致表面电位、离子吸附平衡时离子实际到达位置的电位、电荷密度和电场强度均存在差异;表面电位(绝对值)随着电解质浓度降低而增加,电荷密度随着电解质浓度降低而降低。碱金属离子在蒙脱石-Cu2+表面吸附的离子特异性效应本质上是由活化能决定的。通过本研究建立的离子吸附模型预测的活化能与化学反应中阿累尼乌斯定律所预测结果一致,从而进一步验证了新模型理论的正确性。本研究表明建立的离子-表面相互作用模型在固/液界面反应中具有普适性应用,为进一步明确离子-带电表面相互作用机制提供了理论基础。
[1] Kunz W,Henle J,Ninham B W. ‘Zur Lehre von der Wirkung der Salze’(About the science of the effect of salts):Franz Hofmeister's historical papers. Current Opinion in Colloid & Interface Science,2004,9(1/2):19—37.
[2] Gao X D,Li H,Tian R,et al. Quantitative characterization of specific ion effects using an effective charge number based on the Gouy-Chapman model. Acta Physico-Chimica Sinica,2014,30(12):2272—2282. [高晓丹,李航,田锐,等. 利用基于Gouy-Chapman模型的离子有效电荷定量表征离子特异性效应. 物理化学学报,2014,30(12):2272—2282.]
[3] Kunz W,Lo Nostro P,Ninham B W. The present state of affairs with Hofmeister effects. Current Opinion in Colloid & Interface Science,2004,9(1/2):1—18.
[4] Ninham B W. Physical chemistry:The loss of certainty//Lipid and Polymer-Lipid Systems. Berlin,Heidelberg:Springer Berlin Heidelberg,2002:1—12.
[5] Tobias D J,Hemminger J C. Chemistry:Getting specific about specific ion effects. Science,2008,319(5867):1197—1198.
[6] Lo Nostro P,Ninham B W. Hofmeister phenomena:An update on ion specificity in biology. Chemical Reviews,2012,112(4):2286—2322.
[7] Liu X M,Li H,Li R,et al. Strong non-classical induction forces in ion-surface interactions:General origin of Hofmeister effects. Scientific Reports,2015,4:Article number 5047.
[8] Du W,Li R,Liu X M,et al.Specific ion effects on ion exchange kinetics in charged clay. Colloids and Surfaces A:Physicochemical and Engineering Aspects,2016,509:427—432.
[9] Du W,Li R,Liu X M,et al. Estimating Hofmeister energy in ion-clay mineral interactions from the Gouy-Chapman theory. Applied Clay Science,2017,146:122—130.
[10] Li H,Hou J,Liu X M,et al. Combined determination of specific surface area and surface charge properties of charged particles from a single experiment. Soil Science Society of America Journal,2011,75(6):2128—2135.
[11] Liu X M,Li H,Du W,et al. Hofmeister effects on cation exchange equilibrium:Quantification of ion exchange selectivity. The Journal of Physical Chemistry C,2013,117(12):6245—6251.
[12] Wu Y B,Liu X M,Li R,et al. Hofmeister effect in ion adsorption kinetics on surface of yellow earth particles. Acta Pedologica Sinica,2018,55(6):1450—1459. [吴英博,刘新敏,李睿,等. 黄壤颗粒表面离子吸附动力学中的离子特异性效应. 土壤学报,2018,55(6):1450—1459.]
[13] Huang C Y,Xu J M. Soil science. 3rd ed. Beijing:China Agriculture Press,2011. [黄昌勇,徐建明. 土壤学. 3版. 北京:中国农业出版社,2011.]
[14] Li H,Wu L S,Zhu H L,et al. Ion diffusion in the time-dependent potential of the dynamic electric double layer. Journal of Physical Chemistry C,2009,113(30):13241—13248.
[15] Li R,Li H,Zhu H L,et al. Kinetics of cation adsorption on charged soil mineral as strong electrostatic force presence or absence. Journal of Soils and Sediments,2011,11(1):53—61.
[16] Li R,Li H,Liu X M,et al. Combined measurement of surface properties of particles and equilibrium parameters of cation exchange from a single kinetic experiment. RSC Advances,2014,4(47):24671—24678.
[17] Li Y T,Liu X M,Tian R,et al. An approach to estimate the activation energy of cation exchange adsorption. Acta Physico-Chimica Sinica,2017,33(10):1998—2003. [李嫣婷,刘新敏,田锐,等. 一种评估阳离子交换吸附活化能的方法. 物理化学学报,2017,33(10):1998—2003.]
[18] Liu X M,Li H,Li R,et al. Generalized Poisson—boltzmann equation taking into account ionic interaction and steric effects. Communications in Theoretical Physics,2012,58(3):437—440.
[19] Eriksson E. Cation-exchange equilibria on clay minerals. Soil Science,1952,74(2):103—114.
[20] Li R,Li H,Xu C Y,et al. Analytical models for describing cation adsorption/desorption kinetics as considering the electrostatic field from surface charges of particles. Colloids and Surfaces A:Physicochemical and Engineering Aspects,2011,392(1):55—66.
[21] Zarzycki P,Thomas F. Theoretical study of the acid–base properties of the montmorillonite/electrolyte interface:Influence of the surface heterogeneity and ionic strength on the potentiometric titration curves. Journal of Colloid and Interface Science,2006,302(2):547—559.
[22] Liu H Y,Liu X M,Tian R,et al. Specificeffect of aggregating montmorillonite nanoparticles. Acta Pedologica Sinica,2018,55(3):673—682. [刘汉燚,刘新敏,田锐,等. 蒙脱石纳米颗粒聚集中的离子特异性效应. 土壤学报,2018,55(3):673—682.]
[23] Tian R,Yang G,Li H,et al. Activation energies of colloidal particle aggregation:Towards a quantitative characterization of specific ion effects. Physical Chemistry Chemical Physics,2014,16(19):8828—8836.
[24] Parsons D F,Boström M,Lo Nostro P,et al. Hofmeister effects:interplay of hydration,nonelectrostatic potentials,and ion size. Physical Chemistry Chemical Physics,2011,13(27):12352—12367.
[25] Liu X M,Hu F N,Ding W Q,et al. A how-to approach for estimation of surface/Stern potentials considering ionic size and polarization. The Analyst,2015,140(21):7217—7224.
[26] Li Q Y,Tang Y,He X H,et al. Approach to theoretical estimation of the activation energy of particle aggregation taking ionic nonclassic polarization into account. AIP Advances,2015,5(10):Article number 107218.
[27] Brown M A,Goel A,Abbas Z. Effect of electrolyte concentration on the Stern layer thickness at a charged interface. Angewandte Chemie International Edition,2016,55(11):3790—3794.
[28] Wu C H,Li L,Lei C,et al. Research and application of silicate passivation agent in remediation of heavy metal-contaminated soil:A review. Soils,2017,49(3):446—452. [武成辉,李亮,雷畅,等. 硅酸盐钝化剂在土壤重金属污染修复中的研究与应用. 土壤,2017,49(3):446—452.]
Specificity of Alkali Metal Ions Absorbed on Surface of Montmorillonite-Cu2+
PAN Xiaoli, LIU Xinmin, LI Hang, LI Rui†
(Chongqing Key Laboratory of Soil Multi-scale Interfacial Process, College of Resources and Environment, Southwest University, Chongqing 400715, China)
The interactions between ions and charged particles determine microscopic properties of particles, interface reaction processes and interactions between particles. Ion exchange adsorption is an important physicochemical process of the reactions at solid/liquid interfaces. In this study, based on the newly established ion adsorption kinetics model, adsorption kinetics of alkali metal ions on the surface of montmorillonite particles (a surface with permanent charges) was characterized, in an attempt to analyze specificity of the ions in the adsorption process and to provide theoretical support to studies on interactions between ions and charged particle surface.Kinetics of the adsorption of alkali metal ions, Li+, Na+and K+on the surface of montmorillonite-Cu2+particles was studied with the aid of the constant flow method, relative to ion concentration of the soluttion. Relationship between ion equilibrium adsorption capacity and system activation energy in 1︰1 electrolyte (LiNO3, NaNO3, KNO3) solution was established.(1) Li+, Na+and K+varied sharply in adsorption rate and equilibrium adsorption capacity, in solutions the same in concentration, and in the ions adsorption process, they exhibited apparent ion specificities. Adsorption selectivities of the ions were subject to concentration of the electrolyte, and in solutions, 1×10–4mol·L–1and 1×10–3mol·L–1in electrolyte concentration, the metal ions exhibited an order of K+>> Na+> Li+in equilibrium adsorption capacity, while in solutions, 1×10–2mol·L–1in electrolyte concentration, they did an order of K+>> Li+> Na+, which suggests that volume of the ions is a major factor affecting equilibrium adsorption capacity in solutions high in electrolyte concentration. The adsorption processes of Li+, Na+and K+appeared to be of first-order kinetics as affeted by weak force. Their desorption processes did too, but with apparent ion specificities; (2) In the same electrolyte system,(the distance between alkali metal ions and clay mineral surface when the adsorption reaches equilibrium) decreased with increasing electrolyte concentration, exhibiting an order ofNa>Li>Kin solutions high in electrolyte concentration (1×10–2mol·L–1) , and an order ofLi>Na>Kin solutions low in electrolyte concentration (1×10–4mol·L–1and 1×10–3mol·L–1) . Obviouslyof K+is always the lowest regardless of electrolyte concentration because it has a layer of softer outer electron cloud around, and hence is much stronger in non-classical polarization than Na+and Li+, but for Na+and Li+, in solutions high in electrolyte concentration volume might be a main factor affecting ion adsorption processes, thus resulting inNa>Li, while in solutions low in electrolyte concentration, non-classical polarization would play a leading role, weakening the volume effect, and reversing the order asLi>Na. Therefore the joint effect of non-classical polarization and volume effect of the ions determines position of the ions in the double electric layer and then equilibrium adsorption capacity, thus leading to differences in surface potential (absolute value) , which increases with decreasing electrolyte concentration, exhibiting an order of Li+>Na+>K+in solutions the same in concentration, which indicates that surface potential is mainly influenced by non-classical polarization; and (3) As the newly established model can be used to predict positions of the ions in the double electric layer, and further to calcuate activation energy of the system. The activation energy decreases with increasing electrolyte concentration, and in solutions regardless of eletrolyte concentration and type of cation, both adsorption saturation and activation energy of cations follow a similar law.The occurrence of ion specificities is caused and determined mainly by activation energy. All the findings in this study demonstrate that the newly established model of cation exchange adsorption is universally applicable to researches on solid/liquid interface reaction.
Charged surface; Ion adsorption; Surface potential; Non-classical polarization; Activation energy; Ion specific effects
S153.3
A
10.11766/trxb201904110079
潘小丽,刘新敏,李航,李睿. 碱金属离子在蒙脱石-Cu2+表面吸附的离子特异性[J]. 土壤学报,2020,57(2):370–380.
PAN Xiaoli,LIU Xinmin,LI Hang,LI Rui.Specificity of Alkali Metal Ions Absorbed on Surface of Montmorillonite-Cu2+. [J]. Acta Pedologica Sinica,2020,57(2):370–380.
* 国家自然科学基金项目(41530855,41877026)和重庆市自然科学基金项目(cstc2018jcyjAX0318)资助 Supported by the National Natural Science Foundation of China(Nos.41530855 and 41877026)and the Natural Science Foundation of Chongqing in China(No. cstc2018jcyjAX0318)
,E-mail:lirui02282005@126.com
潘小丽(1993—),女,重庆人,硕士研究生,主要从事土壤表面电化学研究。E-mail: 773259511@qq.com
2019–04–11;
2019–06–19;
优先数字出版日期(www.cnki.net):2019–07–18
(责任编辑:陈荣府)
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国药学药品知识仓库(2022年1期)2022-03-23
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2021年1期)2021-04-13
中国畜牧杂志(2021年1期)2021-01-20
华声文萃(2019年1期)2019-09-10
中学生理科应试(2019年3期)2019-07-08
考试周刊(2016年66期)2016-09-22
中学化学(2015年5期)2015-07-13
中学化学(2015年5期)2015-07-13
