
基于机器学习和大数据挖掘的药物重定位算法综述
2020-04-18 10:46彭超胡永祥陈龙飞叶紫璇田埂
药学进展 2020年1期
彭超,胡永祥,陈龙飞,叶紫璇,田埂*
(1. 湖南工业大学计算机学院,湖南 株洲 412007; 2. 元码基因科技(北京)股份有限公司,北京 100102)
药物重定位(又称为药物重新使用或药物重新配置)是将现有药物应用于新的疾病的过程[1]。与传统的药物研发方法相比,药物重定位可以显著降低成本。药物重新定位的一个显著优势是,由于重新定位的药物已经通过了大量的安全测试,因此它的安全性是已知的,从而降低了药物研发失败的风险。此外,重新定位的药物可以节省将药物推向市场所需的早期成本和时间,从而加快了从基础研究工作到临床治疗的过渡。德国会计律师事务所(Deloitte & Touche )于2016 年发布的一份研究报告显示医药研发巨头公司的投资回报率从2010 年的10.1%下降到了2016 年的3.7%。同时,研发一种新药的平均成本从不足12 亿美元增加到15.4 亿美元,研发时间需要14 年[2]。Nosengo 等[3]得出如下结论:目前新的药物进入市场需要13 ~ 15 年,耗费资金在20 ~ 30 亿美元之间,并且成本还在不断上升。一些调查结果显示,重新定位药物成本平均只有3 亿美元,进入市场大约需要6.5 年。
药物重定位主要包括基于机器学习的方法、大数据挖掘定位的方法和基于活体定位的方法。基于机器学习和大数据挖掘的药物重定位方法依赖于治疗后细胞株的基因表达反应,或者依赖于药物与疾病之间的多层次信息关系,并且利用公共数据库和生物信息学工具系统地识别药物与蛋白靶点之间的相互作用网络。由于几十年来蛋白质与药效之间结构信息的积累,该方法已逐渐取得成功,与基于活体方法相比,基于机器学习和大数据挖掘的药物再定位技术具有速度快、成本低等优点。基于机器学习和大数据挖掘的药物再定位技术已成为一项潜在的强大技术。
本文介绍了近年来计算药物重定位的研究进展。重点介绍基于特征的方法、基于矩阵分解的方法和基于网络的方法。
1 基于特征的方法
基于计算方法的药物重定位的方法利用公共数据库和生物信息学工具系统地确定药物和目标蛋白之间的相互作用网络。 但是高分辨率的靶点结构信息、 疾病表型信息或药物基因表达谱会增加特征数据集的维度。例如:美国癌细胞系百科全书项目(cancer cell line encyclopedia,CCLE)研究了5 万多个特征表示上千万个基因的mRNA 表达和突变状态。用于训练的样本数量明显少于可用特征的数量,所有这些特征的直接应用都会导致模型过拟合,而实际上,只有一小部分特征集对药物敏感性预测有作用。因此,研究人员提出了基于特征的方法。基于特征的方法主要分为基于传统机器学习算法的方法和基于深度学习的方法。
1.1 基于传统机器学习算法的方法
机器学习算法与药物-标靶相互作用网络信息结合,为药物研发提供了新思路。2006 年,Guengerich[4]利用机器学习算法揭示了P450 酶在药物代谢和毒性中所产生的作用。Napolitano 等[5]将非线性支持向量机(support vector machines,SVM)应用于药物的疗效分类上。Gottlieb 等[6]利用逻辑回归算法对药物进行重定位。Yabuuchi 等[7]将药物的化学描述信息与靶蛋白序列组合为混合特征矩阵,并利用SVM 预测新的蛋白靶标。Gönen[8]利用机器学习中的贝叶斯算法对药物与靶蛋白进行预测,寻找新的药物与靶蛋白关联关系。基于机器学习的药物重定位模型如图1 所示。首先将药物与副作用信息、药物化学结构信息和疾病与基因的相关信息进行整合,然后通过特征提取和特征选择得到训练数据。选择相关机器学习算法进行训练,最后利用训练好的算法模型得到药物重定位结果。
特征提取方法将原始特征投影到一个新的维数较低的特征空间中,新得到的特征通常是原始特征的组合,目的是发现更多有意义的信息。特征提取技术常见的有主成分分析法(principal component analysis,PCA)、奇异值分解法(singular value decomposition,SVD)。
特征选择方法的目的是根据一些设计标准从完整的输入特征集中选择一小部分特征,作为模型的输入。在预测药物敏感性的过程中,通常将先验生物学知识纳入特征部分。例如:基于路径的弹性网络正则化,它将路径整合到以数据驱动的特征选择中。基于生物学通路的特征选择,将信号和调控通路与基因表达数据相结合,选择具有最低冗余的重要特征或利用信号通路的激活状态作为特征。常见的特征选择方法有过滤式、包裹式和嵌入式法。常用的特征选择方法以及它们的特点如表1 所示。
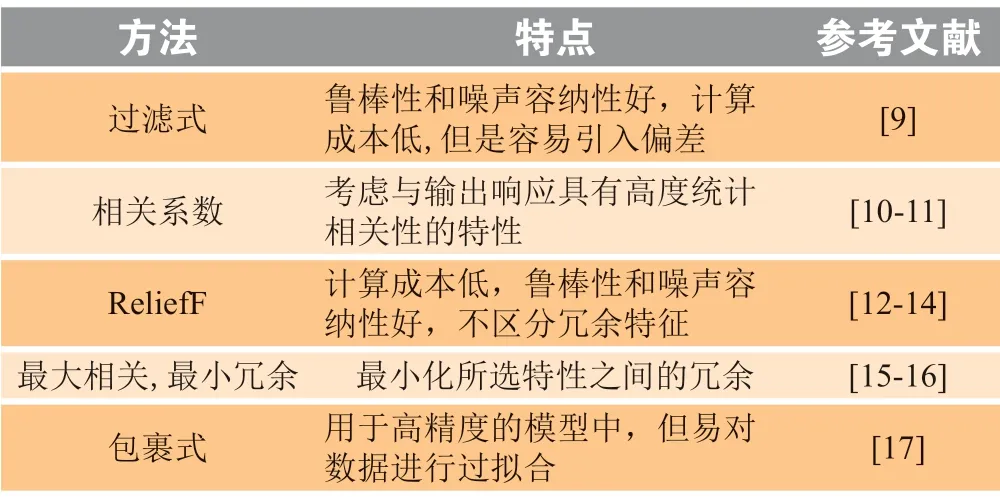
表 1 特征选择方法Table 1 Feature selection methods
利用传统机器学习算法对特定药物进行重定位可以提高药物定位结果预测的准确性,降低研发成本,缩短研发时间。但是,随着大数据时代的到来,传统的机器学习方法逐渐变得难以适应复杂的样本,由于存在对复杂函数的表示能力有限、学习能力不强等不足,它们往往只能提取初级特征。同时,因为以人工方式选取特征的步骤繁复冗杂,传统的机器学习方法有时并不能有效地挖掘数据中蕴含的丰富信息。
1.2 基于深度学习的方法
深度学习算法是机器学习算法的新方向,其本质是深层次的神经网络。深度学习通过模拟人脑建立计算模型,具有强大的自动提取特征的能力以及有效的特征表征能力,能够获取不同层次的信息。基于以上优点,深度学习在药物重定位方面也得到了应用。Korotcov等[18]将深度神经网络(deep neural network,DNN)与其他多种机器学习方法在药物研发的多个方面进行系统比较,结果表明,深度学习的表现优于传统机器学习算法。Rodríguez-Pérez 等[19]构建小分子-靶标的活性谱, 并利用深度学习模型进行测试。Lusci 等[20]将递归神经网络(recurrent neural network,RNN)与化合物的表征方式结合,并得到了很高的准确度。Segler 等[21]基于深度学习结合蒙特卡洛算法的方法简单高效,得到了专业人员的肯定。Hughes 等[22]利用深度学习模型研发了第一个能够对化合物进行快速筛选的模型。Turk 等[23]提取ChEMBL 数据库中匹配分子作为深度学习模型的数据集。
深度学习模型往往需要大量标记样本进行训练,对标记样本的需求很高。在生物医学和药物研发的应用场景下,标记样本的获取依赖于领域专家知识和实验验证,成本较高。同时,基于深度学习得到的模型的端到端的计算模式使得研究者不能理解深度学习模型提取的特征所表征的含义,从而难以在药物研发过程中做出合理可靠的决策。
2 基于矩阵完成的方法
计算药物重新定位的基础之一是准确预测药物-靶点相互作用(drug-target interaction,DTI)。DTI 可以用药物和靶标的二进制标记矩阵Y 表示,如果药物Di和靶标Sj相互作用,则矩阵Y 中的元素Ri, j为1,否则Ri, j为0。预测DTI 的问题也就转化为从Y 的已知元素中估计未知元素的标签的问题。药物-靶标关联矩阵Y如下图2 所示。
近年来研究人员提出了各种预测DTI的计算方法。其中,基于贝叶斯的矩阵分解方法被广泛应用于药DTI矩阵,如表2 所示。
矩阵分解能够将较高维度的数据映射为2 个低维度矩阵的乘积,从而能够很好地解决数据的稀疏性问题,并且矩阵分解的具体实现和求解很简洁,便于理解。基于矩阵分解建立的模型的预测准确度较高,具有很强的扩展性,其基本思想能够运用在各种场景中。但是,矩阵分解模型也有一定的局限性。例如:1)模型的可解释性差,其隐藏空间中的维度并没有和药物学中的概念对应;2)模型的训练速度慢,且不能通过离线训练来弥补这个缺点;3)只是单纯的运用数学原理解决问题,并没有将生物、药物中的信息加入模型。

表 2 基于贝叶斯的矩阵分解方法Table 2 Bayesian matrix factorization method
3 基于网络的方法
在过去的十几年中,基于网络的方法已成为预测药物敏感性的最常用方法之一。由于药物开发成本的增加和新批准药物的数量的减少,找出已上市药物的一些新价值变得十分有必要。其中一些方法有助于更恰当地设计独特的药物靶点组合和联合药物治疗,稳健的通道将改善特定患者的治疗。一些学者建议研究药物应用,疾病治疗与基因的关系。一些文献从生物系统和网络结构框架的角度分析了疾病的诊断、治疗和药物发现之间的关系。各种高通量数据的积累使生物分子和细胞网络的重建成为可能。基于网络的方法,通过化学相似性进行相关分析,可以为新药副作用的发现以及已上市药物重定位提供线索。基于网络的药物重定位方法可分为2类:1)基于药物-疾病相似性的方法;2)基于网络相似性推理的方法。
3.1 基于药物-疾病相似性方法
近年来研究人员提出许多基于药物-疾病相似性的药物重定位方法,例如:Guney 等[29]引入了一种药物-疾病相似性度量,该度量可量化药物靶标与疾病之间的相互作用。该方法引入化学相似性进行关联,并且考虑了必要的生物信息,具有很强的系统性和综合性。实验结果表明基于网络的邻近度可以帮助我们量化药物的治疗效果并预测新的药物-疾病关联。Kotlyar 等[30]对药物如何破坏网络,以及基于网络的药物表征会直接影响参与结合的对象进行了总结。他们首次使用网络表征受药物差异调节的基因。李鹏[31]在疾病网络的基础上基于质量作用定律,建立了基于网络扰动动力学模型的分析工具PerturbationAnalyzer。该方法通过整合定量蛋白质组学和蛋白相互作用网络数据,从蛋白质相互作用的浓度依赖关系出发,将蛋白浓度变化对网络扰动程度作为靶标辨识的重要依据。Chen 等[32]构建了一个通用的异构网络,该网络包含通过蛋白质-蛋白质序列相似性,药物-药物化学相似性和已知的药DTI 而链接的药物和蛋白质,挖掘潜在的药物-疾病关联。
3.2 基于网络相似性推理方法
很多研究人员把关注点放在网络相似性推理:Cheng 等[33]提出了一种基于网络的推理(network-based inference,NBI)方法,该方法仅使用药物-靶标二分网络拓扑相似性来推断已知药物的新靶标。Chen 等[34]基于Zhou 等[35]开发的推荐技术的推理方法,提出基于网络拓扑度量来预测直接的药物-疾病关联。他们通过挖掘有关药物-疾病两方网络特性的数据,将问题表述为推荐给特定药物的疾病。Wang 等[36]提出了一个基于异构网络模型的计算框架,这种计算框架可以捕获疾病、药物和靶标之间的相互关系,以预测新的药物使用情况。一些学者通过一些特殊案例,例如帕金森病,试图通过定位网络模块来重新定位药物。Yue 等[37]开发了针对失调的药物靶标网络通路或途径而非单个靶点的疗法,并建立了一个将全基因组关联分析数据与帕金森病患者3 个脑区域的基因共表达模块整合在一起的框架。
基于网络相似性推理的方法便于理解,简单可靠,性能优于基于药物-疾病的方法和基于靶点相似性的方法。同时,研究人员可以根据具体研究的需要对网络输出的结果排序,但是基于网络推理的方法只适用在药物靶点关系已知的情况下,因此不能预测新药物的靶点,从而带来了很大的局限性。
4 结语与展望
本文介绍了基于机器学习和大数据挖掘的药物重定位的研究进展。重点介绍基于特征的方法、基于矩阵完成的方法和基于网络的方法。在基于特征的方法中,无论是机器学习还是深度学习对数据的要求都比较高,需要专业人员设计标签,从而增加了药物研发的时间。基于矩阵完成的方法不用人为设定标签,研发时间相对其他方法也有所减少,但是矩阵完成只是单纯的引入数学计算,并没有将药物信息引入计算,可能导致计算结果和实际结果有一定偏差。基于网络推理的方法虽然简单可靠,便于理解,但是不能预测新药物的靶点,局限性很大。
随着大数据挖掘技术的发展,基于机器学习和大数据挖掘算法的药物重定位将为疾病的治疗提供更多更有效的方法,已经成为生物医学研究关注的焦点。有理由相信,理性推理和计算模型将在未来的药物重定位过程中发挥重要作用。另外,随着深度学习中无监督学习技术的发展,标签在深度学习处理海量数据方面的影响也越来越小,深度学习,特别是无监督学习与药物重定位结合将是未来研究的重点。
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
数学物理学报(2022年5期)2022-10-09
环球时报(2022-07-13)2022-07-13
环球时报(2022-03-14)2022-03-14
军民两用技术与产品(2021年10期)2021-03-16
河北画报(2020年8期)2020-10-27
世界农药(2019年3期)2019-09-10
电影(2018年8期)2018-09-21
浙江大学学报(工学版)(2016年2期)2016-06-05
肿瘤影像学(2015年3期)2015-12-09
