
利用WGCNA鉴定棉花抗黄萎病相关基因共表达网络
2020-04-16 09:46傅明川陈义珍柳展基刘任重王立国
作物学报 2020年5期
傅明川 李 浩 陈义珍 柳展基 刘任重 王立国
利用WGCNA鉴定棉花抗黄萎病相关基因共表达网络
傅明川*李 浩 陈义珍 柳展基 刘任重 王立国
山东棉花研究中心 / 农业部黄淮海棉花遗传改良与栽培生理重点实验室, 山东济南 250100
加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)作为一种典型的系统生物学方法, 可用来鉴定协同表达的基因模块, 探索模块与目标性状之间的生物学相关性, 并挖掘网络中的核心基因。黄萎病(wilt)可严重降低棉花(spp.)的产量及品质, 因此研究抗病基因及抗病机制, 对于棉花抗病育种工作具有重要意义。本研究以黄萎病菌侵染不同时间点的海岛棉()幼苗根尖转录组数据为基础, 分析其差异表达基因, 并选取在不同样本之间表达水平变异最大的前50%基因(共35,647个)进行WGCNA。结果表明, 在黄萎病菌侵染条件下, 共有22,850个基因差异表达, 其中4685个为所有侵染时间点共有的差异基因。共鉴定到18个基因共表达模块, 其中5个为抗黄萎病相关特异性模块(black、mediumpurple3、darkolivegreen、plum3模块分别与侵染2 h、6 h、48 h、72 h时间点正相关; mediumpurple2与侵染2 h时间点负相关)。GO和KEGG富集分析表明, 特异性模块可以富集到有生物学意义的富集结果, 如刺激响应、钙离子结合、黄酮类化合物合成代谢等抗逆相关通路。通过计算模块内基因的连通性, 挖掘出了网络中的核心基因, 功能预测表明, 这些基因可能在逆境胁迫响应中发挥重要作用。本研究为进一步理解棉花抗黄萎病的分子机制、选育棉花抗病新品种提供了理论指导。
棉花; 黄萎病; 加权基因共表达网络分析; 核心基因
棉花(spp.)是世界范围内重要的纤维及油料作物之一, 其在我国经济发展中也占有十分重要的地位。黄萎病(wilt)作为土壤传播的维管束系统性病害, 是棉花生产中最严重的病害之一。过去通常认为黄萎病菌的致病机制是“导管堵塞”; 目前研究表明, 黄萎病菌分泌的毒素可能是其主要致病因素, 其中细胞壁降解酶、脂多糖蛋白复合物以及核蛋白可能是重要的致病成分。Wang等[1]研究证实, 黄萎病菌VdNEP蛋白是一种重要的黄萎病致病因子。通过研究高致病性菌株Vd080发现,基因与黄萎病菌的致病力和微菌核形成有关[2]。构成了黄萎病菌的RGS基因家族, 其中在孢子生成、菌丝生长、微菌核形成过程中发挥重要作用[3]。由于缺乏有效的黄萎病防治药剂, 选育和种植抗病品种是目前最经济有效、绿色环保的防治措施。但抗病基因资源缺乏、抗病机制不明, 导致棉花抗黄萎病育种工作进展缓慢[4]。因此, 挖掘棉花抗病基因、探究其抗病机制, 对于棉花抗病育种工作具有重要意义。Kawchuk等[5]利用图位克隆在番茄()位点中分离出2个紧密相连的抗黄萎病主效基因和,研究证实基因可介导感病马铃薯品种()对黄萎病菌的抗性。进一步研究表明, 由于棉花黄萎病菌缺少基因, 导致在棉花中过表达并不能明显增强其抗病性[6]。基因组学、转录组学及蛋白质组学的迅速发展, 为研究棉花抗黄萎病的分子机制提供了有利基础。Zhang等[7]通过对黄萎病菌侵染下的海岛棉()“Pima 90-53”幼苗根系进行转录组分析, 首次报道了“SA→NPR1→TGA→PR-1→抗病”代谢通路。目前, 在棉花中已鉴定出一些抗病相关基因。如是海岛棉中一个ERF转录因子基因, 转的烟草()植株内抗病相关基因表达量升高, 并可提高转基因植株的抗病性[8];为一类丝氨酸/苏氨酸蛋白激酶基因, 参与调控多个抗逆相关信号通路, 从而在抵御病原菌侵染胁迫及氧化应激响应中发挥作用[9];参与调控活性氧含量及JA信号通路相关基因的表达, 转基因的拟南芥()植株对黄萎病菌的抗性提高[10]。此外,[11]、[12]、[13]等基因也均在抗病响应中发挥重要作用。
目前, 棉花基因组测序工作已陆续完成, 这为从海量生物数据中挖掘有用信息提供了坚实基础。相比传统分子生物学方法, 借助生物信息学手段, 可更加快捷地定位目标基因, 挖掘与性状相关的关键基因。加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)以芯片或RNA-seq表达数据为基础, 通过幂指数加权构建无尺度拓扑重叠矩阵, 用以描述基因之间的相互关系, 并依此将表达模式相近的基因划分到一个基因表达模块中[14]。WGCNA多用来研究协同表达的基因模块与目标性状之间的生物学相关性, 并探索基因共表达网络中的核心基因。作为一种典型的系统生物学方法, WGCNA已广泛应用于植物学研究中。如Tan等[15]通过分析镉处理不同时间点的17个水稻 ()转录组数据, 鉴定得到22个基因模块, 结合差异表达分析, 共挖掘到164个镉胁迫响应相关基因; Zou等[16]通过对2个棉花品系不同发育时期的纤维转录组数据进行WGCNA, 共鉴定得到5个纤维发育相关特异性模块, 并挖掘出模块内的核心基因; 通过对14份玉米()不同发育阶段的转录组数据进行WGCNA, 研究者鉴定得到14个组织特异性模块, 并对其中2个模块的基因互作网络进行了进一步研究, 从中挖掘到了、、等开花相关核心基因[17]。
本研究以黄萎病菌侵染不同时间点的棉花转录组数据为材料, 对其进行差异表达分析; 通过构建加权基因共表达网络, 划分基因模块, 并筛选出抗病相关特异性模块; 经GO及KEGG富集分析, 探究模块功能; 根据基因在相应网络中的连通性, 鉴定出模块内的核心基因。本研究可为进一步理解棉花抗黄萎病的分子机制提供理论基础, 并为棉花抗病育种工作提供新的基因资源。
1 材料与方法
1.1 数据获取及差异表达分析
棉花在黄萎病菌侵染不同时间点的转录组数据下载自NCBI数据库(PRJNA234454)[18]。该数据以高抗黄萎病海岛棉品种“海7124”为材料, 对生长2周的幼苗根系使用Vd8孢子悬液侵染。分别选取侵染2、6、12、24、48和72 h的根尖, 提取总RNA, 并以未侵染材料(0 h)为对照。首先, 利用fastq-dump软件(https://ncbi.github.io/sra-tools/fastq-dump.html)将下载得到的SRA文件转换为fastq格式文件, 然后利用FastQC (http://www.bioinformatics.babraham. ac.uk/projects/fastqc/)对测序结果进行质量评估, 并通过Trimmomatic软件[19]进行质控, 将得到的clean data用于后续分析。以海岛棉基因组为参考基因组[20], 使用HISAT2[21]进行序列比对后, 利用featureCounts[22]计数, 得到每个基因在各个样本中的raw counts。将数据导入R中, 利用DESeq2[23]进行差异表达分析。本研究选取|log2FC| > 1 (log2FC代表处理与对照表达量比值的对数值)及adj<0.001 (adj代表校正后的值)的基因作为差异表达基因。
1.2 加权基因共表达网络构建
利用R程序中的WGCNA软件包[14]进行加权基因共表达网络构建。以标准化后的基因表达矩阵做为输入, 共21个转录组样本(7个时间点, 各3次重复)。通过计算每个基因在各个样本间表达水平的变异程度, 选取变异最大的前50%基因进行WGCNA。经过阈值筛选, 最终选择=18对原始有尺度关系矩阵进行幂处理, 得到无尺度化邻接矩阵。为更好评估基因间表达模式的相关性, 进一步将邻接矩阵转化为拓扑重叠矩阵(Topological Overlap Matrix, TOM), 并利用拓扑相异矩阵(dissTOM=1-TOM), 采用动态剪切算法进行基因聚类及模块划分。模块内最少基因数为30 (minModuleSize=30), 相似模块合并阈值为0.25 (cutHeight=0.25), 网络类型为“signed” (type="signed"或networkType="signed", 依不同函数而定)。
1.3 特异性模块筛选
对每个模块中的所有基因进行主成分分析(Principle Component Analysis, PCA), 将主成分1(PC1)的值称为该模块的模块特征向量(Module Eigengene, ME)。为筛选抗病相关特异性模块, 分别计算每个模块的模块特征向量与不同性状之间(此处为不同侵染时间)的相关系数及相应值。>0代表正相关,<0代表负相关。本研究选择||>0.70及<0.001的模块作为特异性模块进一步分析。
1.4 富集分析及代谢通路分析
利用R程序中的clusterProfiler软件包[24]进行GO (Gene Ontology)和KEGG (Kyoto Encyclopedia of Genes and Genomes)富集分析。阈值为<0.01及<0.05。
1.5 转录因子分析
将各模块中的蛋白序列提交到PlantTFDB数据库(http://planttfdb.cbi.pku.edu.cn/prediction.php#)[25]分析预测, 从而得到每个模块中的转录因子。
1.6 RNA提取及qRT-PCR
选取生长2周、长势一致的“海7124”幼苗, 采取浸根法用浓度1×107个mL-1的黄萎病菌Vd8孢子悬液侵染10 min。选取侵染后2、6、12、24、48、72 h和相应时间点未侵染幼苗根系, 利用TRIzol法提取总RNA, 取1 μg RNA反转录为cDNA。
qPCR使用Thermo Fisher Scientific的QuantStudio 5实时荧光定量PCR系统, 选用Aidlab公司的2×Sybr Green qPCR Mix试剂盒, 荧光染料为SYBR Green, 内参基因为。反应程序95℃预变性3 min; 95℃ 15 s, 60℃ 15 s, 40个循环; 熔点曲线程序为95℃ 15 s, 60℃ 1 min, 95℃ 15 s。反应体系包含2×SYBR qPCR Mix 10 μL、DNA Template (稀释10倍) 0.8 μL、正向引物(10 µmol L-1) 0.4 μL、反向引物(10 µmol L-1) 0.4 μL、ddH2O 8.4 μL。使用2–DDCt法分析基因相对表达量[26], 设置3次生物学重复。所用基因引物见表1。
2 结果与分析
2.1 差异表达分析
通过分析棉花在黄萎病菌侵染不同时间点(0 h、2 h、6 h、12 h、24 h、48 h、72 h)的转录组数据, 最终共得到22,850个差异表达基因(|log2FC| > 1,adj< 0.001)。其中, 显著上调基因9398个, 显著下调基因13,171个, 另有281个基因在不同时间点表现出不同的上下调趋势。差异表达基因最多的时间点出现在侵染后24 h (14,679个), 最少的为2 h (10,729个), 所有时间点共有的差异表达基因为4685个(图1)。
2.2 加权基因共表达网络构建
对表达矩阵中变异较低的基因进行过滤, 最终选取35,647个基因进行WGCNA。当=18时, 无尺度网络拟合指数2>0.8, 平均连通性趋近于0, 表明用此值进行幂处理可以得到符合要求的无尺度网络, 因此选择=18构建无尺度网络(图2)。采用动态剪切算法对基因进行聚类及模块划分, 通过计算每个模块的模块特征向量, 合并相似模块, 最终共得到18个基因共表达模块(图3)。模块内基因数量为61 (palevioletred2) ~ 8431 (black)个。各模块内差异表达基因所占比例为11% (thistle2) ~ 81% (paleturquoise)。值得注意的是, 除black及mediumpurple2模块外, 同一模块内的差异表达基因通常表现出相似的上下调模式, 如paleturquoise、turquoise模块内的差异基因均为显著下调, 而palevioletred2、thistle2模块内均为显著上调。
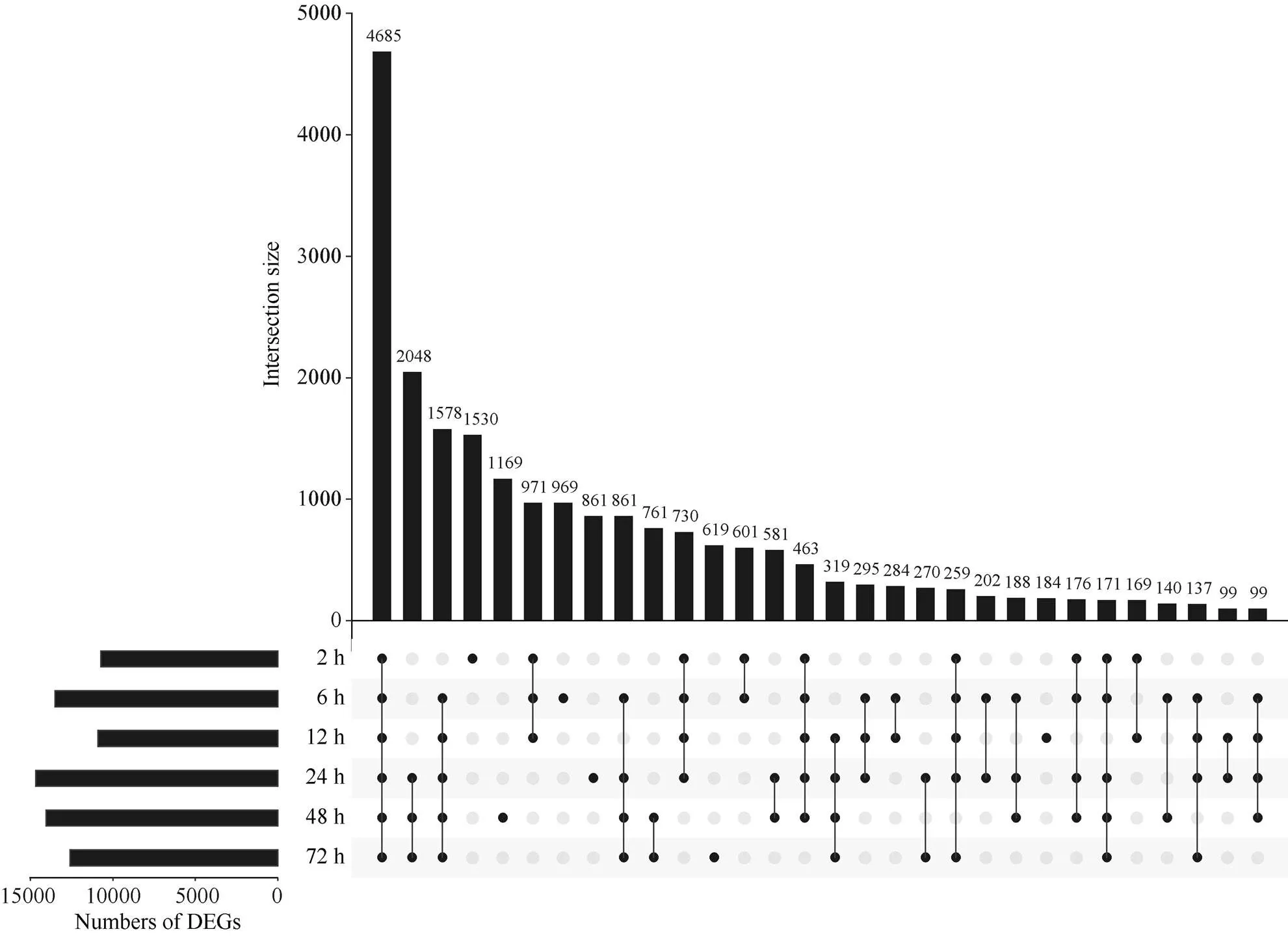
图1 不同侵染时间点的差异表达基因
只有一个黑点的列代表某个数据集特有的差异基因; 有2个或以上用实线连接的黑点的列代表相应数据集之间共有的差异基因。
The column with one black point represents the DEGs only in the corresponding set; the column with two or more black points linked by a solid line represents the intersection among the sets.
转录因子是生物过程中一类重要的调控蛋白, 本研究也对每个模块内的转录因子进行了进一步分析。结果表明, 模块内转录因子数量分布为3 (mediumpurple2) ~ 590 (black)个, 在相应模块内所占比例为2% (mediumpurple2) ~ 19% (plum3), 不同模块内转录因子的分布类型存在差异, 但主要集中在ERF (9.27%)、MYB (8.09%)、bHLH (7.94%)、WRKY (7.03%)、C2H2 (5.74%)、NAC (5.36%)、bZIP (5.28%)等转录因子家族, 报道表明这些转录因子均参与调控植物的抗逆过程[27]。每个模块内差异表达的转录因子数占该模块内所有转录因子的比例为20% (thistle2) ~ 89% (navajowhite1), 数量显著偏高(χ2=92.24,=2e-12), 进一步表明转录因子在抗逆调控过程中可能发挥重要作用。

图2 软阈值的选择
a: 不同软阈值下的无尺度网络拟合指数(2), 红线代表2=0.8。b: 不同软阈值下的平均连通性。
a: scale-free topology fit index as a function of the soft-thresholding power, the red line represents that2is equal to 0.8. b: mean connectivity as a function of the soft-thresholding power.

图3 基因聚类树及模块划分
a: 基于拓扑相异矩阵构建的基因聚类树。b: 使用动态剪切算法得到的基因模块, 不同颜色代表不同模块。c: 合并相似模块后的模块划分结果。
a: gene clustering on TOM-based dissimilarity. b: module division by dynamic tree cut, different colors represent different modules. c: module division after merging similar modules.
2.3 抗病相关特异性模块鉴定
在18个基因模块中, 有5个与黄萎病菌侵染存在高度特异性(||>0.70,<0.001)。其中, black (=0.72,=4e-04)、mediumpurple3 (=0.78,=5e-05)、darkolivegreen (=0.76,=9e-05)、plum3 (=0.97,=6e-12)模块分别与侵染2 h、6 h、48 h、72 h时间点正相关; mediumpurple2 (=-0.82,=8e-06)与侵染2 h时间点负相关(图4)。
2.4 特异性模块富集分析
GO通路可分为生物过程(Biological Process, BP)、分子功能(Molecular Function, MF)及细胞组分(Cellular Component, CC) 3类。分析结果表明, black和mediumpurple3模块分别富集到41个和36个生物过程, 其中black模块主要富集到氮化合物转运(GO:0071705)、蛋白定位(GO:0045184)、蛋白转运(GO:0015031)等调控通路, 以及一些信号相关通路,如信号转导(GO:0007165)、信号转导调控(GO:1902531)等(图5-a); mediumpurple3模块主要富集到一些代谢相关通路, 如氨基酸代谢(GO:0006520)、硫化物代谢(GO:0006790)、甘氨酸代谢(GO:0006544)等生物学过程(附表1)。此外, black、mediumpurple3、darkolivegreen、plum3及mediumpurple2模块分别富集到21、7、7、1和2个分子功能, 如蛋白质转运蛋白活性(GO:0008565)、钙离子结合(GO:0005509)(附表2); black和mediumpurple2模块分别富集到44和4个细胞组分, 最显著的分别为膜衣(GO:0030117)和蛋白酶体核心复合物(GO:0005839)(附表3)。

图4 模块与性状相关性热图
每行代表一个模块, 每列代表一种性状。矩形框里的数字代表模块与性状之间的相关系数及相应值。抗病相关特异性模块用红色标示, 其基因及转录因子数量标于左侧模块矩形框内。
Each row corresponds to a module, and each column corresponds to a trait. The correlation coefficient and the correspondingvalue are shown in each cell. The specific modules which significantly associated withinfection are colored in red, and the corresponding numbers of genes and transcription factors are shown in the left cells.
KEGG富集分析表明, black、mediumpurple3和darkolivegreen模块分别富集到8、10和1个KEGG代谢通路, 其中black模块主要富集到mRNA监测通路、内吞作用和蛋白质输出等通路(图5-b); mediumpurple3主要富集到氨基酸生物合成、柠檬酸循环和碳代谢等通路; darkolivegreen富集到黄酮类化合物生物合成代谢通路(附表4)。

图5 Black模块的GO和KEGG富集分析
纵坐标代表GO条目或KEGG代谢通路, 横坐标代表富集到的基因在模块内所占比率。点的大小代表富集到的基因数量, 点的颜色代表多重校验后的值大小。
The vertical axis represents the enriched GO term or KEGG pathway, and the horizontal axis represents the ratio of enriched genes in the module. The point size represents the gene number enriched in the pathway, and the color represents the-value corrected for multiple testing.
2.5 核心基因鉴定及基因互作网络构建
核心基因通常指模块内具有高连通性的基因, 本研究选取每个模块中kME值(Eigengene Connectivity)最高的前5个基因做为核心基因, 并利用核心基因及其互作基因绘制基因互作网络图(图6)。通过与拟南芥进行同源比对, 对核心基因及其互作基因进行了功能注释(表2, 附表5)。此外, 本研究选取了9个核心基因进行了qRT-PCR分析, 结果表明其表达模式与转录组数据基本一致(图7)。
3 讨论
本研究以黄萎病菌侵染下海岛棉幼苗根系RNA-seq数据为基础, 对其进行了差异表达分析, 共鉴定出22,850个差异表达基因。Chen等[18]利用该转录组数据共得到17,517个差异表达基因, 结果产生差异的主要原因是在分析过程中使用了不同的参考基因组及分析软件。Chen等[18]以雷蒙德氏棉()基因组为参考基因组, 比对率约为76%, 本研究以海岛棉基因组为参考基因组, 比对率约为94%, 因此比对效果更好。Zhang等[28]通过分析黄萎病菌侵染24 h下陆地棉()幼苗根的转录组数据, 共鉴定到4794个差异表达基因, 其中一些基因在本研究中具有相似的差异表达模式(如、、等), 但也有部分基因在2个研究中表现出相反的上下调变化趋势(如、、等), 暗示这些基因在海岛棉和陆地棉的抗病过程中可能发挥不同作用。
通过WGCNA方法, 本研究共鉴定到5个(black、mediumpurple3、darkolivegreen、plum3、mediumpurple2)抗病相关特异性模块。富集分析结果表明, 特异性模块可得到具有相关生物学意义的功能富集和代谢通路富集结果。如GO富集分析中, black模块可富集到刺激响应(GO:0051716)、激素介导的信号通路(GO:0009755)、激素刺激应答(GO:0032870)等抗逆相关细胞过程; plum3模块富集到钙离子结合分子功能(GO:0005509), 而研究表明钙离子是植物防御应答过程中一类非常重要的第二信使[29]; KEGG富集分析中, darkolivegreen模块可富集到黄酮类化合物生物合成代谢通路, 此类化合物是植物中所特有的一类多功能复合物, 在植株抵御生物/非生物胁迫过程中发挥重要作用[30]。
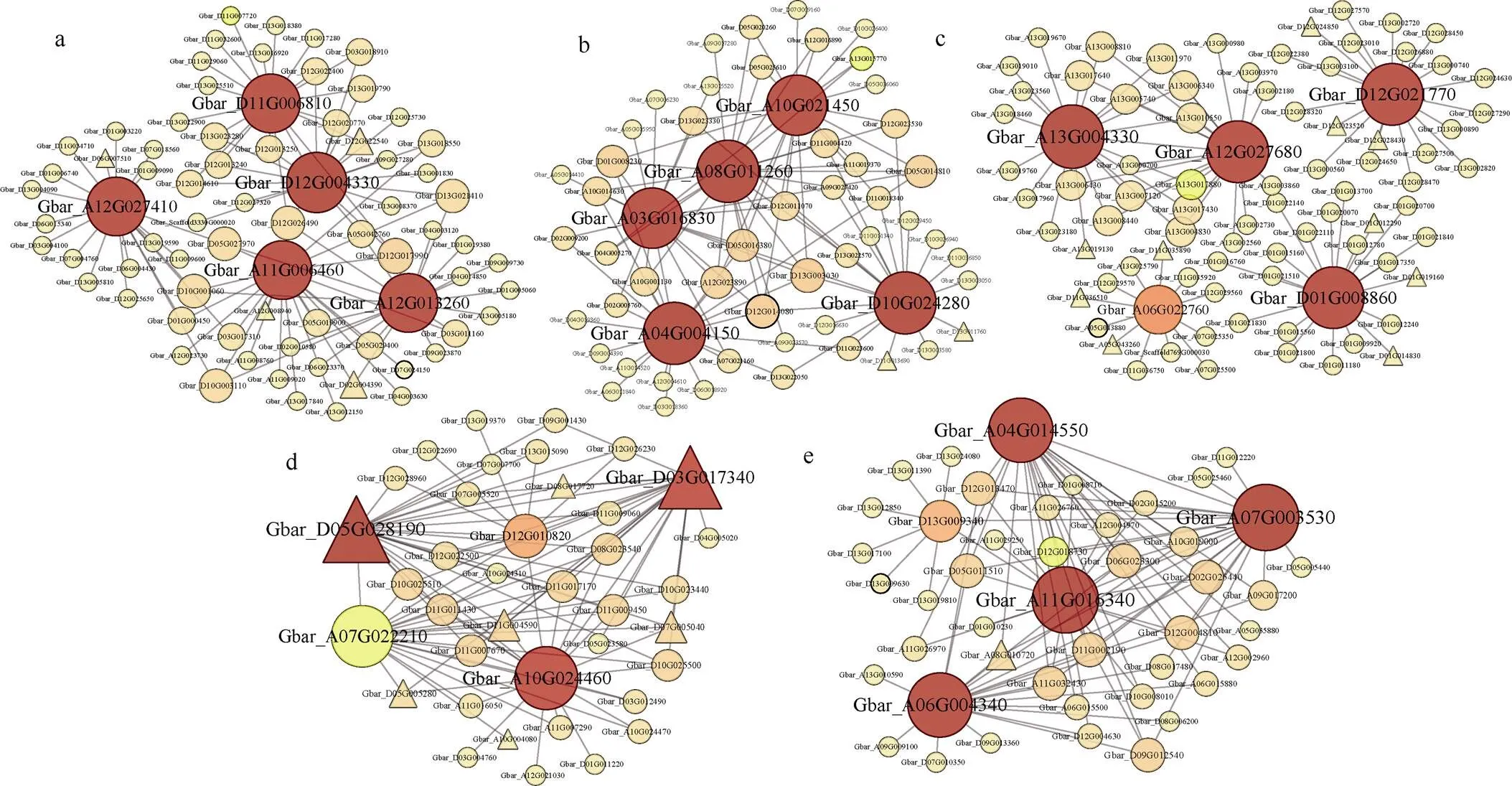
图6 抗病相关特异性模块内的基因共表达网络
点的大小和深浅代表该基因在网络中连通性的高低。转录因子用三角形表示, 其他基因用圆形表示。a: Black模块内的基因共表达网络。b: Mediumpurple3模块内的基因共表达网络。c: Darkolivegreen模块内的基因共表达网络。d: Plum3模块内的基因共表达网络。e: Mediumpurple2模块内的基因共表达网络。
The genes with higher connectivity in the corresponding network are shown with larger circle sizes and darker colors. The transcription factors are indicated by triangles, with other genes being indicated by circles. a: Gene co-expression network of the black module. b: Gene co-expression network of the mediumpurple3 module. c: Gene co-expression network of the darkolivegreen module. d: Gene co-expression network of the plum3 module. e: Gene co-expression network of the mediumpurple2 module.

图7 核心基因表达分析
a: qRT-PCR分析, 上方垂直线代表3次重复的标准差。b: RNA-seq表达热图。
a: expression analysis by qRT-PCR, the bars indicate standard deviation of three replications. b: heat map of the hub genes.
*:< 0.05, **:< 0.01.

表2 抗病相关特异性模块中核心基因的功能注释
通过计算模块内基因的连通性, 可推测该基因在网络中的位置及重要程度。结合已经报道过的棉花中抗黄萎病相关基因发现, 这些基因在相应模块内都具有较高的连通性。如black模块中的基因(, kME=0.89), 可通过激活基因的表达介导棉花抗病过程[31]; mediumpurple2模块中的(, kME=0.89)为一类NBS-LRR (nucleotide-binding site–leucine rich repeat)基因, 在棉花抗黄萎病侵染过程中发挥重要作用[32]。此外, darkolivegreen模块中的、plum3模块中的在相应模块内都具有较高连通性,其在陆地棉中的同源基因及均为抗病相关基因[33-34]。其他非抗病相关特异性模块中也包含一些高连通性的抗病基因, 如turquoise模块中的(, kME= 0.90)[35]、(, kME=0.95)[36]及mediumorchid模块中的(, kME=0.91)[37]基因等。
选取特异性模块中连通性最高的前5个基因为核心基因, 推测其可能在抗病过程中发挥重要作用。这些基因在棉花中的功能大多尚不明确, 而它们在拟南芥中的同源基因部分已报道为抗逆相关基因。如plum3模块中的为一个ERF转录因子家族基因, 其在拟南芥中的同源基因为, 该基因可通过调控JA/ET信号通路响应病原菌胁迫[38]; darkolivegreen模块中的同源基因参与调控ABA信号转导, 在植株非生物胁迫应答中发挥作用[39]; black模块中在拟南芥中的同源基因为, 该基因通过激活G6PD蛋白调节细胞内氧化还原反应平衡, 从而在植株响应逆境胁迫中发挥重要作用[40]。
与核心基因连接度较高的基因, 同样可能在逆境胁迫应答中发挥作用。如在darkolivegreen模块中,的高连接度基因在拟南芥中的同源基因为, 该基因可通过调节活性氧含量、细胞死亡过程及基因的表达参与响应病原菌侵染胁迫[41]; 在plum3模块中,的高连接度基因在拟南芥中的同源基因编码一个细胞分裂素信号通路相关转录因子ARR12, 参与调控植株的干旱胁迫应答[42]; 在black模块中,的高连接度基因在拟南芥中的同源基因编码PAD4蛋白, 可与另一个抗病相关蛋白EDS1组成复合体共同调控SA信号通路, 进而在病原菌胁迫应答中发挥作用[43]。此外, 结合差异表达分析结果表明, 以上基因基本都发生了显著上下调, 从而在表达水平上也说明它们可能在抗病过程中发挥作用。
本研究构建的网络中核心基因在棉花中的具体生物学功能目前大多尚不明确, 后续可进一步通过过量表达、VIGS、基因敲除等生物技术手段对其开展深入研究。
4 结论
通过构建权重基因共表达网络共鉴定到5个抗病相关特异性模块, 并揭示了它们的生物学意义。以连通性为指标, 揭示了特异性模块中可能在抗病过程中发挥重要作用的关键基因。本研究结果为进一步理解棉花抗病过程的分子机制提供了理论指导。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb. aspx。
[1] Wang J Y, Cai Y, Gou J Y, Mao Y B, Xu Y H, Jiang W H, Chen X Y. VdNEP, an elicitor from, induces cotton plant wilting., 2004, 70: 4989–4995.
[2] Li Z F, Liu Y J, Feng Z L, Feng H J, Klosterman S J, Zhou F F, Zhao L H, Shi Y Q, Zhu H Q., encoding CYC8 glucose repression mediator protein, is required for microsclerotia formation and full virulence in, 2015, 10: e0144020.
[3] Xu J, Wang X Y, Li Y Q, Zeng J G, Wang G L, Deng C Y, Guo W Z. Host-induced gene silencing of a regulator of G protein signaling gene () confers resistance towilt in cotton, 2018, 16: 1629–1643.
[4] 朱荷琴, 李志芳, 冯自力, 冯鸿杰, 魏锋, 赵丽红, 师勇强, 刘世超, 周京龙. 我国棉花黄萎病研究十年回顾及展望棉花学报, 2017, 29(增刊): 37–50. Zhu H Q, Li Z F, Feng Z L, Feng H J, Wei F, Zhao L H, Shi Y Q, Liu S C, Zhou J L. Overview of cottonwilt research over the past decade in china and its prospect in future., 2017, 29(suppl): 37–50 (in Chinese with English abstract).
[5] Kawchuk L M, Hachey J, Lynch D R, Kulcsar F, van Rooijen G, Waterer D R, Robertson A, Kokko E, Byers R, Howard R J, Fischer R, Prüfer D. Tomatodisease resistance genes encode cell surface-like receptors, 2001, 98: 6511–6515.
[6] 刘琳琳, 张文文, 周易, 苗玉焕, 许莲, 刘敏, 张坤, 张献龙, 朱龙付. 棉花与番茄抗棉花黄萎病不依赖于中国科学: 生命科学, 2014, 44: 803–814. Liu L L, Zhang W W, Zhou Y, Miao Y H, Xu L, Liu M, Zhang K, Zhang X L, Zhu L F. Resistance of cotton and tomato tofrom cotton is independent on., 2014, 44: 803–814 (in Chinese).
[7] Zhang Y, Wang X F, Ding Z G, Ma Q, Zhang G R, Zhang S L, Li Z K, Wu L Q, Zhang G Y, Ma Z Y. Transcriptome profiling ofinoculated withprovides a resource for cotton improvement, 2013, 14: 637.
[8] Zuo K J, Qin J, Zhao J Y, Ling H, Zhang L D, Cao Y F, Tang K X. Over-expression GbERF2 transcription factor in tobacco enhances brown spots disease resistance by activating expression of downstream genes, 2007, 391: 80–90.
[9] Zhang Y, Wang X F, Li Y Y, Wu L Z, Zhou H M, Zhang G Y, Ma Z Y. Ectopic expression of a novel Ser/Thr protein kinase from cotton (), enhances resistance toinfection and oxidative stress in, 2013, 32: 1703–1713.
[10] Li T G, Wang B L, Yin C M, Zhang D D, Wang D, Song J, Zhou L, Kong Z Q, Klosterman S J, Li J J, Adamu S, Liu T L, Subbarao K V, Chen J Y, Dai X F. TheTIR-NBS-LRR genemediates resistance againstwilt, 2019, 20: 857–876.
[11] Munis M F H, Tu L L, Deng F L, Tan J F, Xu L, Xu S C, Long L, Zhang X L. A thaumatin-like protein gene involved in cotton fiber secondary cell wall development enhances resistance againstand other stresses in transgenic tobacco, 2010, 393: 38–44.
[12] Mo H J, Wang X F, Zhang Y, Zhang G Y, Zhang J F, Ma Z Y. Cotton polyamine oxidase is required for spermine and camalexin signalling in the defence response to, 2015, 83: 962–975.
[13] He X, Zhu L F, Wassan G M, Wang Y J, Miao Y H, Shaban M, Hu H Y, Sun H, Zhang X L. GhJAZ2 attenuates cotton resistance to biotic stresses via the inhibition of the transcriptional activity of GhbHLH171, 2018, 19: 896–908.
[14] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis, 2008, 9: 559.
[15] Tan M P, Cheng D, Yang Y N, Zhang G Q, Qin M J, Chen J, Chen Y H, Jiang M Y. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes, 2017, 17: 194.
[16] Zou X Y, Liu A Y, Zhang Z, Ge Q, Fan S M, Gong W K, Li J W, Gong J W, Shi Y Z, Tian B M, Wang Y L, Liu R X, Lei K, Zhang Q, Jiang X, Feng Y L, Zhang S Y, Jia T T, Zhang L P, Yuan Y L, Shang H H. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton () using RNA sequencing analysis, 2019, 10: 119.
[17] 杨宇昕, 桑志勤, 许诚, 代文双, 邹枨. 利用WGCNA进行玉米花期基因共表达模块鉴定作物学报, 2019, 45: 161–174. Yang Y X, Sang Z Q, Xu C, Dai W S, Zou C. Identification of maize flowering gene co-expression modules by WGCNA., 2019, 45: 161–174 (in Chinese with English abstract).
[18] Chen J Y, Huang J Q, Li N Y, Ma X F, Wang J L, Liu C, Liu Y F, Liang Y, Bao Y M, Dai X F. Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response towilt, 2015, 15: 148.
[19] Bolger A M, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data, 2014, 30: 2114–2120.
[20] Wang M J, Tu L L, Yuan D J, Zhu D, Shen C, Li J Y, Liu F Y, Pei L L, Wang P C, Zhao G N, Ye Z X, Huang H, Yan F L, Ma Y Z, Zhang L, Liu M, You J Q, Yang Y C, Liu Z P, Huang F, Li B Q, Qiu P, Zhang Q H, Zhu L F, Jin S X, Yang X Y, Min L, Li G L, Chen L L, Zheng H K, Lindsey K, Lin Z X, Udall J A, Zhang X L. Reference genome sequences of two cultivated allotetraploid cottons,and, 2019, 51: 224–229.
[21] Kim D, Langmead B, Salzberg S L. HISAT: a fast spliced aligner with low memory requirements, 2015, 12: 357.
[22] Liao Y, Smyth G K, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features, 2014, 30: 923–930.
[23] Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, 2014, 15: 550.
[24] Yu G C, Wang L G, Han Y Y, He Q Y. clusterProfiler: an R package for comparing biological themes among gene clusters, 2012, 16: 284–287.
[25] Jin J P, Tian F, Yang D C, Meng Y Q, Kong L, Luo J C, Gao G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants, 2016, 45: D1040–D1045.
[26] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCTmethod, 2001, 25: 402–408.
[27] Erpen L, Devi H S, Grosser J W, Dutt M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants, 2018, 132: 1–25.
[28] Zhang W W, Zhang H C, Liu K, Jian G L, Qi F J, Si N. Large-scale identification ofgenes associated withby comparative transcriptomic and reverse genetics analysis, 2017, 12: e0181609.
[29] Lecourieux D, Ranjeva R, Pugin A. Calcium in plant defence-signalling pathways, 2006, 171: 249–269.
[30] Liu J Y, Osbourn A, Ma P D. MYB transcription factors as regulators of phenylpropanoid metabolism in plants, 2015, 8: 689–708.
[31] Li C, He X, Luo X Y, Xu L, Liu L L, Min L, Jin L, Zhu L F, Zhang X L. Cotton WRKY1 mediates the plant defense- to-development transition during infection of cotton byby activatingexpression, 2014, 166: 2179–2194.
[32] Yang J, Ma Q, Zhang Y, Wang X F, Zhang G Y, Ma Z Y. Molecular cloning and functional analysis of, a gene inthat plays an important role in conferring resistance towilt, 2016, 575: 687–694.
[33] Yang J, Wang G N, Ke H F, Zhang Y, Ji L L, Huang L Z, Zhang C Y, Wang X F, Ma Z Y. Genome-wide identification of cyclophilin genes inand functional characterization of a CYP with antifungal activity against, 2019, 19: 272.
[34] Xiong X P, Sun S C, Li Y J, Zhang X Y, Sun J, Xue F. The cotton WRKY transcription factornegatively regulates the defense response against, 2019, 7: 393–402.
[35] Cheng H Q, Han L B, Yang C L, Wu X M, Zhong N Q, Wu J H, Wang F X, Wang H Y, Xia G X. The cotton MYB108 forms a positive feedback regulation loop with CML11 and participates in the defense response againstinfection, 2016, 67: 1935–1950.
[36] Li Y B, Han L B, Wang H Y, Zhang J, Sun S T, Feng D Q, Yang C L, Sun Y D, Zhong N Q, Xia G X. The thioredoxin GbNRX1 plays a crucial role in homeostasis of apoplastic reactive oxygen species in response toinfection in cotton, 2016, 170: 2392–2406.
[37] Duan X P, Zhang Z D, Wang J, Zuo K J. Characterization of a novel cotton subtilase genein response to extracellular stimulations and its role inresistance, 2016, 11: e0153988.
[38] Moffat C S, Ingle R A, Wathugala D L, Saunders N J, Knight H, Knight M R. ERF5 and ERF6 play redundant roles as positive regulators of JA/Et-mediated defense againstin, 2012, 7: e35995.
[39] Kumar D, Kumar R, Baek D W, Hyun T K, Chung W S, Yun D J, Kim J Y.functions as a positive regulator in plant responses to ABA, 2017, 10: 223–243.
[40] Dal Santo S, Stampfl H, Krasensky J, Kempa S, Gibon Y, Petutschnig E, Rozhon W, Heuck A, Clausen T, Jonak C. Stress-induced GSK3 regulates the redox stress response by phosphorylating glucose-6-phosphate dehydrogenase in, 2012, 24: 3380–3392.
[41] Pan Q N, Cui B M, Deng F Y, Quan J L, Loake G J, Shan W X.encodes a novel endoplasmic reticulum (ER)-localized protein inand negatively regulates resistance against biotrophic pathogens, 2016, 209: 1641–1654.
[42] Nguyen K H, Ha C V, Nishiyama R, Watanabe Y, Leyva-González M A, Fujita Y, Tran U T, Li W Q, Tanaka M, Seki M, Schaller G E, Herrera-Estrella L, Tran L-S P.type B cytokinin response regulators ARR1, ARR10, and ARR12 negatively regulate plant responses to drought, 2016, 113: 3090–3095.
[43] Cui H T, Qiu J D, Zhou Y, Bhandari D D, Zhao C H, Bautor J, Parker J E. Antagonism of transcription factor MYC2 by EDS1/PAD4 complexes bolsters salicylic acid defense ineffector-triggered immunity, 2018, 11: 1053–1066.
Identification of co-expressed modules of cotton genes responding toinfection by WGCNA
FU Ming-Chuan*, LI Hao, CHEN Yi-Zhen, LIU Zhan-Ji, LIU Ren-Zhong, and WANG Li-Guo
1Cotton Research Center of Shandong Academy of Agricultural Sciences / Key Laboratory of Cotton Breeding and Cultivation in Huang-Huai-Hai Plain, Ministry of Agriculture, Jinan 250100, Shandong, China
Weighted gene co-expression network analysis (WGCNA) is a classic systematic biological method, which can be used to identify co-expressed modules, investigate relationships between modules and specific traits, and screen hub genes in the networks.wilt, caused by the fungal pathogen, can cause severe fibre quality reduction and yield loss of cotton. Studying on cotton genes and molecular mechanisms related to defense responses againstcan shed light on cotton breeding. In this study, 21 transcriptome data ofseedling roots infected bywere used to investigate differentially expressed genes (DEGs). By filtering out the genes with low variation, 35,647 genes were selected for WGCNA. In total, 22,850 DEGs were identified underinfection, with 4685 in common at all inoculated time points. Co-expression network analysis identified 18 modules, in which five modules significantly associated withinfection (black, mediumpurple3, darkolivegreen, plum3 positively correlated with the 2 h, 6 h, 48 h, and 72 h inoculated time points, respectively; mediumpurple2 negatively correlated with the 2 h inoculated time point). GO and KEGG enrichment analysis were performed on these five specific modules, which could be enriched in GO terms and metabolic pathways, such as cellular response to stimulus, calcium ion binding and flavonoid biosynthesis. The hub genes were screened by calculating gene connectivity in the corresponding networks, and they may play important roles in the resistance against biotic/abiotic stresses. In summary, by WGCNA, a few defense-related co-expressed modules and hub genes were identified. The results of this study would be beneficial for further understanding of the molecular mechanisms of pathogen resistance in cotton, and provide new gene resources for cotton breeding in the future.
cotton;wilt; WGCNA; hub gene
10.3724/SP.J.1006.2020.94124
本研究由山东省农业科学院农业科技创新工程(CXGC2018B01, CXGC2018E06)项目资助。
This study was supported by the Agricultural Science and Technology Innovation Project of Shandong Academy of Agricultural Sciences (CXGC2018B01, CXGC2018E06).
2019-08-23;
2020-01-15;
2020-02-18.
傅明川, E-mail: fumingchuan@shandong.cn
URL: http://kns.cnki.net/kcms/detail/11.1809.s.20200218.1558.002.html
猜你喜欢
作物学报(2022年9期)2022-07-18
中国农业科学(2022年11期)2022-06-27
今日农业(2022年4期)2022-06-01
安徽农业科学(2022年1期)2022-02-14
农业科技与信息(2021年4期)2021-12-05
新农村(浙江)(2021年2期)2021-11-30
江苏农业学报(2021年2期)2021-06-30
发明与创新·大科技(2019年5期)2019-07-31
湖北农业科学(2017年10期)2017-06-22
农家科技下旬刊(2016年6期)2016-05-14
