
Therapy-related acute promyelocytic leukemia with FMS-like tyrosine kinase 3-internal tandem duplication mutation in solitary bone plasmacytoma:A case report
2020-04-09 08:07:54LiLiHongXianFuShengHaiFengZhuang
World Journal of Clinical Cases 2020年19期
Li-Li Hong,Xian-Fu Sheng,Hai-Feng Zhuang
Li-Li Hong,Xian-Fu Sheng,Hai-Feng Zhuang,Department of Hematology,The First Affiliated Hospital of Zhejiang Chinese Medical University,Hangzhou 310000,Zhejiang Province,China
Abstract BACKGROUND Therapy-related acute promyelocytic leukemia(t-APL)is a rare complication observed in solitary bone plasmacytoma(SBP),and SBP after radiotherapy evolving to APL harboring the FMS-like tyrosine kinase 3-internal tandem duplication(FLT3-ITD)mutation has never been reported.Here,we present the first case reported until now.CASE SUMMARY We describe a 64-year-old woman who presented with lumbar pain and was initially diagnosed with SBP.However,after one year of radiotherapy treatment,this patient experienced a long-standing bone-marrow-suppressive period and finally developed APL harboring the FLT3-ITD mutation,as confirmed by analyses of clinical features,bone marrow morphology,flow cytometry,cytogenetic examination,and molecular biology.On admission,the patient had disseminated intravascular coagulation and intracranial hemorrhage,and the peripheral blood and bone marrow smear displayed abundant abnormal promyelocytes.Unfortunately,she died when the definite diagnosis was made.CONCLUSION The patient with t-APL harboring FLT3-ITD mutation evolving from SBP after radiotherapy had not been reported and had poor clinical outcomes.FLT3-ITD mutation in t-APL may be a potential pathogenesis of leukemogenesis.We should consider the potential risk of secondary neoplasms in SBP patients after radiotherapy.
Key Words:Solitary bone plasmacytoma;Therapy-related acute promyelocytic;Leukemia;FMS-like tyrosine kinase 3-internal tandem duplication mutation;Radiotherapy;Cytopenia;Disseminated intravascular coagulation;Case report
INTRODUCTION
Solitary bone plasmacytoma(SBP)is a rare plasma cell myeloma(PCM)presenting as a single bone lesion,the biopsy of which shows infiltration by plasma cells,with no or minimal bone marrow plasmacytosis and an absence of end-organ damage,including anemia,hypercalcemia,and renal dysfunction.It accounts for less than 5% of PCMs[1].Patients presenting with SBP,especially those with detectable plasmacytosis in the bone marrow,have a higher risk for progression to symptomatic multiple myeloma(MM):Approximately 50% of patients with SBP develop MM within 10 years after the initial diagnosis.Radiotherapy is the optimal treatment for SBP,which is highly radiosensitive.Local control rates can be achieved in 80%-90% of patients with radiation therapy alone.A subset of patients may require surgical intervention.Adjuvant chemotherapy is not indicated[2].
Acute promyelocytic leukemia(APL),as defined in the World Health Organization Classification,is a neoplasm with t(15;17)(q22;q21)leading to the formation of a fusion gene,PML/RARα,and a striking response to all-trans retinoic acid(ATRA)therapy;thus,APL has a relatively good prognosis compared with other types of acute myeloid leukemia(AML)[3].Although the complete remission(CR)rate was significantly improved with the ATRA-based regimen,the incidence of early death,mainly due to hemorrhagic complications,is still approximately 20%[4].Up to 12% of all cases of acute promyelocytic leukemia are t-APL[5].The entity was first recognized morphologically in 1970 and was described cytogenetically in the 1980s[6].t-APL was defined as a well-recognized disorder developing after chemotherapy,radiotherapy,immunosuppressive agents,or a combination of these treatments given for a primary disease[7].
SBP after radiotherapy evolving to APL harboring the FMS-like tyrosine kinase 3-internal tandem duplication(FLT3-ITD)mutation,but without progression to MM,has not been reported.Here,we present the first case of t-APL with the FLT3-ITD mutation after SBP radiotherapy.
CASE PRESENTATION
Chief complaints
A 64-year-old woman was admitted to the hematology department of our hospital in August 2019 with pancytopenia complicated by headache.
History of present illness
The patient had recurrent pancytopenia for half a year.The symptom of headache had lasted for 4 d.
History of past illness
The patient presented with a 3-mo history of lumbar pain and was finally diagnosed with solitary bone plasmacytoma a year ago.She had received only radiotherapy,at a dose of 45 Gy,22 times.She was first admitted to the hospital in February 2019 due to persistent pancytopenia.The initial complete blood count(CBC)showed a hemoglobin(HB)level of 96 g/L,white blood cell count(WBC)of 2.5 × 109/L,and platelet(PLT)count of 38 × 109/L.Immunofixation electrophoresis showed that the M component was an IgG-kappa light chain.On bone marrow(BM)aspiration,0.13% of monoclonal plasma cells were observed by flow cytometry(FCM).Biopsy revealed hypoplastic marrow without megakaryocytes.No bone lesion other than primary local lumbar injury was observed in a series of X-ray scans.The patient was diagnosed with SBP with minimal marrow involvement,according to the diagnostic criteria for plasma cell dyscrasia,with concomitant myelosuppression.She underwent treatment with thalidomide,granulocyte colony stimulating factor(G-CSF),and thrombopoietin(TPO),blood transfusion,and other supportive treatments.Peripheral blood was significantly improved after 3 mo of treatment and almost restored to normal.
Physical examination
On admission,the patient was lucid and slurred in speech.The skin mucous membrane of the whole body had scattered haemorrhage spots.Limb activity was normal,and nervous system examination showed no abnormality.
Laboratory examinations
The CBC revealed pancytopenia(HB 70 g/L,WBC 3.0 × 109/L,and PLT 38 × 109/L).A peripheral blood smear showed 16% of abnormal promyelocytes with dendritic protrusions(Figure 1A).Thrombin function test showed that prothrombin and partial thromboplastin times were prolonged,fibrinogen was decreased,and the D-dimers were elevated to 4.5 μg/mL.BM examination was conducted,followed by cytogenetic and molecular analyses using BM specimens.The BM aspirate showed 90% of abnormal promyelocytes with densely packed large granules(Figure 1B),which on flow cytometric immunophenotyping expressed CD13,CD33,CD117,CD56,CD81,and myeloperoxidase;partially expressed CD38 and CD34;and slightly expressed CD15,CD11b,and CD16 HLA-DR(Figure 2).Chromosome analysis using a BM sample revealed a karyotype of 46,XX,t(15;17)(q22;q21)in 20 metaphase cells examined(Figure 3).Reverse transcription-PCR(RT-PCR)analyses confirmed the presence of Sform PML/RARα gene rearrangement.The FLT3-ITD gene mutation was detected by Sanger sequencing(Figure 4).Plasma cells were not detected.A monoclonal peak was continuously observed on serum IFE,showing IgG and kappa-type monoclonal gammopathy.
Personal and family history
The patient was married and had two daughters and one son.No additional family history was obtained.
FINAL DIAGNOSIS
The final diagnosis of the patient presented here was t-APL with the FLT3-ITD mutation.
TREATMENT
Transfusion and other supportive treatments.
OUTCOME AND FOLLOW-UP
Unfortunately,the patient died 2 d after admission due to disseminated intravascular coagulation(DIC)and intracranial hemorrhage.
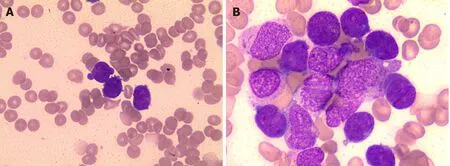
Figure 1 Microscopic examination of peripheral blood and bone marrow.A:PB showed rouleaux formation of red blood cells and an atypical promyelocyte;B:BM aspiration smears showed increased number of atypical promyelocytes with bilobed nuclei and densely packed large granules in the cytoplasm(Wright-Giemsa staining,× 1000).PB:Peripheral blood;BM:Bone marrow.
DISCUSSION
The term t-APL is merely descriptive and a well-recognized complication,based upon a patient’s history of exposure to chemo-and/or radiotherapy.Primary diseases include caners and non-malignant disorders such as multiple sclerosis.The reported latency period ranges from a few months to several years,depending on a number of factors including type of preceding chemotherapy and/or radiation therapy and cumulative therapeutic dose[7,8].Several studies showed no significant differences betweende novoAPL and t-APL in morphological subsets,immunophenotypes of abnormal promyelocytes,cytogenetic abnormalities,and type of PML/RARα fusion gene[5,9].Only a few t-APL cases have a myelodysplastic or pancytopenic preleukemic phase[10].The common manifestation of t-APL includes mucocutaneous bleeding and DIC;the CR rate is approximately 80%,and relapse is uncommon,similar tode novoAPL[7,11].Our case described here points to a peculiar and rapidly fatal form of APL characterized by the presence of FLT3-ITD mutation.To the best of our knowledge,this is the first case report of a patient with SBP after radiotherapy,undergoing a long period of bone marrow suppression,which developed to APL harboring the FLT3-ITD gene mutation.
FMS-like tyrosine kinase 3(FLT3)is a class III receptor tyrosine kinase that is normally expressed in human hematopoietic stem and progenitor cells but is also detected in AML,B-precursor cell acute lymphoblastic leukemia(ALL),a type of T-cell ALL,and chronic myelogenous leukemia(CML)in lymphoid blast crisis.Internal tandem duplication(ITD)and tyrosine kinase domain(TKD)are two distinct forms of FLT3 mutation thought to be involved in leukemogenesis[12,13].FLT3-ITD is the most frequently mutated gene in APL,and the frequencies range from 20.3% to 36.8%[14-17].As many cases of APL exhibit FLT3 mutations,the frequent identification of FLT3 mutations in t-APL is to be expected.AML patients with FLT3 mutations have a poor prognosis,whereas in APL,the presence of an FLT3 mutation seems to result in more severe outcomes[18,19].A meta-analysis showed that the FLT3-ITD mutation had a negative effect on overall survival(OS)and disease-free survival(DFS)in APL.Even when treated with specific drugs such as ATRA and arsenic trioxide,20%-30% of patients still relapse or fail to achieve molecular remission[19].In view of the above,the occurrence of FLT3-ITD mutation in t-APL is considered an adverse prognostic factor.In the case reported here,the patient presented with acute bleeding onset and had a poor outcome.
The potential molecular mechanisms of leukemogenesis in t-APL remain undefined.Studies on leukemogenesis suggest a multistep pathogenesis for t-APL.An agent causes DNA damage,leading to the acquisition of additional mutations that cooperate with the chimeric fusion protein generated by the t(15;17)translocation to induce leukemic transformation;alternatively,an agent modulates the immune system and evades immune surveillance,making the clinically occult,usually trivial,naturally occurring mutations more likely[8].On the one hand,our case of t-APL with FLT3 mutation occurred in SBP after radiotherapy treatment.There was no evidence of dissemination to MM,implying the relationship between radiotherapy and t-APL.On the other hand,the patient in this case presented with long-standing cytopenia concomitant with monoclonal plasma cells on repeated bone marrow analyses.However,the clone of plasma cells disappeared when abnormal promyelocytes were detected.We speculated that one possibility was that the SBP was transformed into APL through a process of clonal evolution.Collectively,the potential pathogenesis of t-APL might be associated with radiotherapy-induced bone marrow suppression,leading to difficulty in hematopoietic recovery,increased clonal evolution changes in hematopoietic stem cells,and accelerated loss of immune surveillance.More importantly,the high frequency of FLT3 mutation was associated with a poor prognosis,which has been considered one of many second-hit events in leukemogenesis and leading to t-APL.In addition,this patient took thalidomide for nearly 6 mo.It has been warned that thalidomide increases the risk of secondary hematological tumors.However,no literature has reported the malignant potential of thalidomide.A meta-analysis indicated that second primary malignancies were reported in MM patients receiving lenalidomide maintenance[20].

Figure 2 Flow cytometric immunophenotyping showing that the bone marrow expressed CD13,CD33,CD117,CD56,CD81,and myeloperoxidase;partially expressed CD38 and CD34;and slightly expressed CD15,CD11b,and CD16.
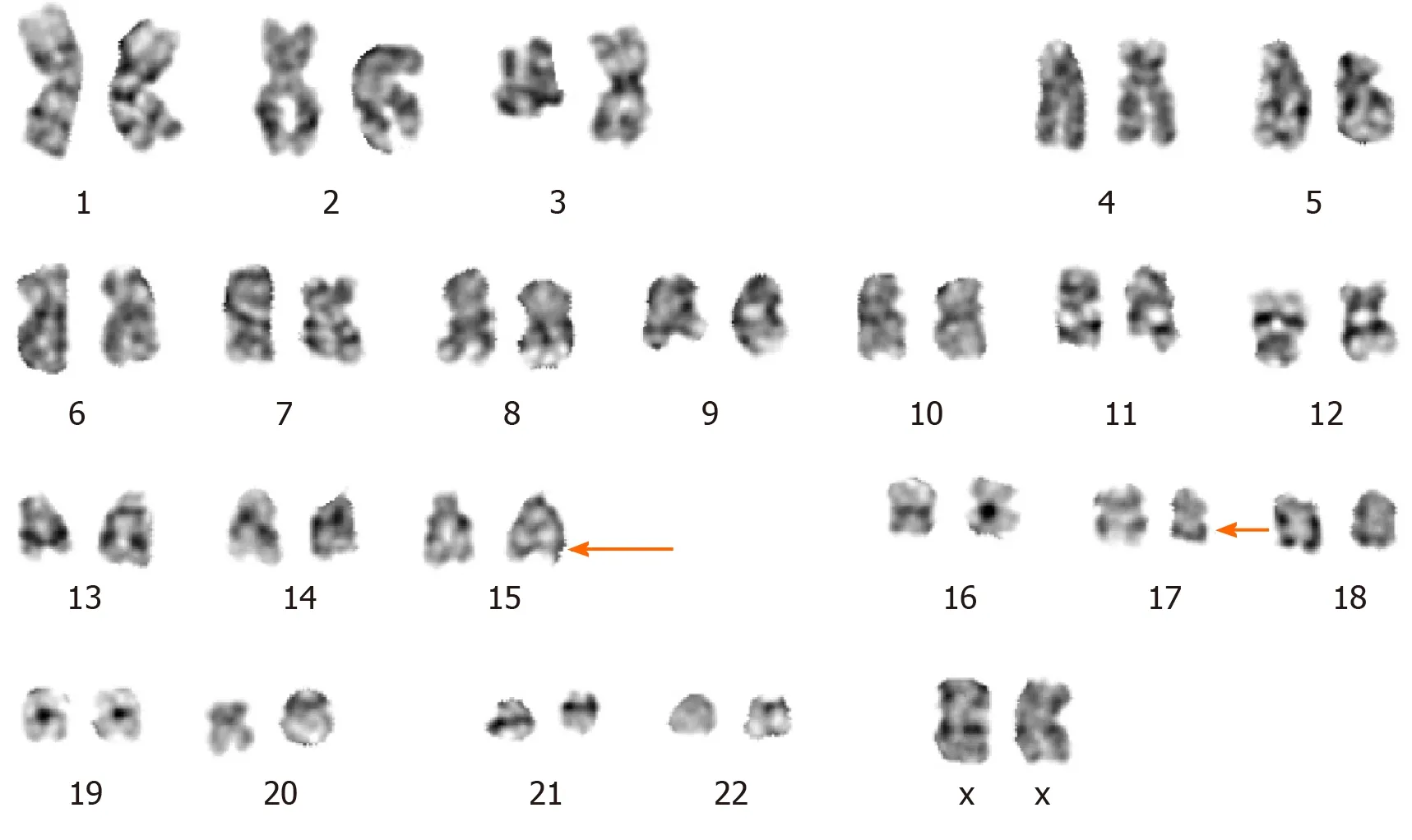
Figure 3 Cytogenetic analysis.G-band karyotyping of bone marrow cells revealed 46,XX,t(15;17)(q22;q21).
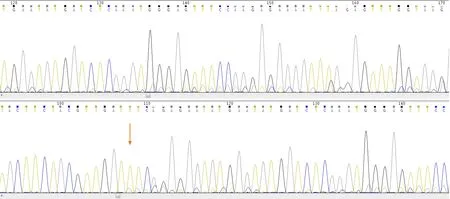
Figure 4 Sanger sequencing results indicating a positive FMS-like tyrosine kinase 3-internal tandem duplication mutation(the orange arrow indicates the mutation site).
The limitation of the study is that SBP and APL came from two different clones,while it is not clear whether t-APL was evolved from SBP.It seems reasonable to propose that the progression to APL after SBP is most likely an outcome of a multifactorial process:The activation of leukemia initiating factors,such as chemotherapy or radiotherapy agents;clone expansion of leukemia stem cells and the acquisition of novel gene mutations;and immune escape resulting from chemotherapy or radiotherapy.Further studies will be needed to accurately elucidate the pathogenesis of APL with FLT3 mutation after SBP.More prospective studies are needed to determine whether the FLT3-ITD mutation status can be incorporated into risk-adapted therapy algorithms.
CONCLUSION
Patients with t-APL secondary to SBP are very rare,and those harboring the FLT3-ITD gene mutation have a worse prognosis thande novoAPL.Our case indicates a possible role for FLT3-ITD mutation as a risk factor for t-APL and may be one of the many second-hit events in leukemogenesis of t-APL.It brings to our attention that we should be alert to the occurrence of a second tumor in patients with SBP after radiotherapy especially with long-term bone marrow suppression.
 World Journal of Clinical Cases2020年19期
World Journal of Clinical Cases2020年19期
- World Journal of Clinical Cases的其它文章
- Parathyroid adenoma combined with a rib tumor as the primary disease:A case report
- Displacement of peritoneal end of a shunt tube to pleural cavity:A case report
- Localized primary gastric amyloidosis:Three case reports
- Bochdalek hernia masquerading as severe acute pancreatitis during the third trimester of pregnancy:A case report
- Intravesically instilled gemcitabine-induced lung injury in a patient with invasive urothelial carcinoma:A case report
- Intraosseous venous malformation of the maxilla after enucleation of a hemophilic pseudotumor:A case report