
Genetic diagnosis history and osteoarticular phenotype of a nontransfusion secondary hemochromatosis
2020-04-07 06:17DanDanRuanYuMianGanTaoLuXiaoYangYaoBinZhuQingHuaYuLiShengLiaoNingLinXinQianJieWeiLuoFaQiangTang
World Journal of Clinical Cases 2020年23期
Dan-Dan Ruan, Yu-Mian Gan, Tao Lu, Xiao Yang, Yao-Bin Zhu, Qing-Hua Yu, Li-Sheng Liao, Ning Lin, Xin Qian, Jie-Wei Luo, Fa-Qiang Tang
Dan-Dan Ruan, Yu-Mian Gan, Tao Lu, Qing-Hua Yu, Li-Sheng Liao, Ning Lin, Xin Qian, Jie-Wei Luo,Fa-Qiang Tang, Shengli Clinical Medical College, Fujian Medical University, Fuzhou 350001,Fujian Province, China
Xiao Yang, Department of Management, Fujian Health College, Fuzhou 350101, Fujian Province, China
Yao-Bin Zhu, Department of Traditional Chinese Medicine, The First Affiliated Hospital, Fujian Medical University, Fuzhou 350005, Fujian Province, China
Fa-Qiang Tang, Department of Orthopedics, Fujian Provincial Hospital, Fuzhou 350001, Fujian Province, China
Abstract BACKGROUND It is not easy to identify the cause of various iron overload diseases because the phenotypes overlap. Therefore, it is important to perform genetic testing to determine the genetic background of patients.AIM To investigate the genetic background of a patient with hemochromatosis complicated by psoriasis on both lower extremities.METHODS Ten years ago, a 61-year-old male presented with iron overload, jaundice,hemolytic anemia and microcytic hypochromic anemia. Computed tomography of the left knee joint showed enlargement of the tibial medullary cavity and thinned bone cortices. Magnetic resonance imaging showed hepatic hemochromatosis,extensive abnormal signals from bone marrow cavities and nodular lesions in the lateral medullary cavity of the upper left lateral tibia. Single photon emission computed tomography showed radial dots of abnormal concentration in the upper end of the left tibia and radial symmetry of abnormal concentrations in joints of the extremities. The patient showed several hot spot mutations of the HFE and G6PD genes detected by next-generation sequencing, but no responsible gene mutation was found. The thalassemia gene was detected by gap-PCR.RESULTS The patient was found to carry the -α4.2 and --SEA deletion mutations of the globin gene. These two mutations are common causes of Southeast Asian α-thalassemia,but rarely cause severe widespread non-transfusion secondary hemochromatosis osteoarthropathy. The simultaneous presence of an auxiliary superposition effect of a rare missense mutation of the PIEZO1 gene (NM_001142864, c.C4748T,p.A1583V) was considered. Moreover, several rare mutations of the IFIH1, KRT8,POFUT1, FLG, KRT2, and TGM5 genes may be involved in the pathogenesis of psoriasis.CONCLUSION The selection of genetic detection methods for hemochromatosis still needs to be based on an in-depth study of the clinical manifestations of the disease.
Key Words: Hemochromatosis; Hemochromatosis osteoarthropathy; Next-generation sequencing; Thalassemia; Gap-PCR; PIEZO1 gene
INTRODUCTION
Hemochromatosis (HC) is an iron overload disease caused by iron metabolism disorders. Large amounts of iron are deposited in the liver, pancreas, heart, joints and other organs, which can cause various degrees of cell destruction, leading to extensive tissue fibrosis, and organ function and structural damage. HC can be divided into two major categories of primary and secondary in terms of etiology. Primary HC is hereditary hemochromatosis (HH), also known as idiopathic HC, caused by a congenital iron metabolism disorder. It is the most common hereditary disease in the Nordic Caucasian population, and is particularly common in Celtic bloodlines such as Ireland, Scotland and Wales. The incidence in non-Hispanic whites in the United States is 1/300[1,2], and the incidence in the northwestern European bloodline can reach 1/150[3,4]. In 1996, the homeostatic iron regulator gene (HFE) was identified as the pathogenic gene in most cases of HH, which encodes a membrane protein that is similar to MHC class I-type proteins, it binds to the transferrin receptor (TFR) and regulates the liver to secrete hepcidin together with iron regulators such as hemojuvelin (HJV) and erythroferrone[5]. TFR1 is expressed in most tissues, including erythroid precursors, liver and myocardium, while TFR2 is expressed only in the liver and intestine. The affinity of TFR1 for iron is about 25 times higher than that of TFR2.This may lead to the rate of iron deposition in different tissue organs[6]. The common responsible mutations ofHFEare: C282Y/C282Y, C282Y/H63D and C282Y/S65C.Non-HFE-related HC is rare, accounting for less than 15% of hereditary iron overload[7], the involved mutant genes are:TFR2,HJV, hepcidin gene (HAMP), which has an autosomal recessive inheritance and ferroportin gene (SLC40A1), rare ferritin regulatory gene, solute carrier family 11 member 2 gene (SLC11A2), which has autosomal dominant inheritance[6,8]. However, there are fewer reports on primary or secondary HC in China. Due to its low incidence and atypical symptoms, it is easy to misdiagnosis and miss the diagnosis. Therefore, we present our study of the genetic background of a patient with HC complicated by psoriasis on both lower extremities.
MATERIALS AND METHODS
Case presentation
In December 2010, a 61-year-old male from a remote mountain area in Fujian, China presented with pain in the left lower extremity and limited activity for one week on admission, and was highly suspected of having bone metastasis from an unknown primary focus. There was no obvious cause of the patient’s pain and swelling in the left knee joint in the morning, walking was limited, and the pain symptoms were alleviated after rest. No significant change in skin color or temperature of both lower limbs was observed after onset. Computed tomography (CT) and X-ray of the left knee joint in hospital showed enlargement of the tibial medullary cavity, thinned cortical bone, and a high-density shadow in the left upper tibia, which was considered to be a bone neoplasm, the nature of which was to be determined (Figure 1B and D). Routine urine analysis showed ERY +++, PRO ++, and LEU negative. Past history showed that 20 years before admission, the patient had an exploratory laparotomy for acute abdomen in a local hospital, but the cause was not determined. The incision failed to effectively heal, resulting in a long-term intestinal mass swelling at the incision which formed an abdominal hernia (Figure 1A). The patient also had psoriasis on both lower extremities (Figure 1C). He had a smoking history for more than 30 years(approximately 20 cigarettes/day), no drinking, no history of blood transfusion, no history of venereal disease and no relevant family history. Physical examination showed that systemic skin color was dark, a 10 cm surgical scar 3 cm from left side of the umbilicus, a soft intestinal mass in the scar the size of a fist could be pushed into the abdomen. Diffuse erythema and scales in both lower extremities, which manifested as erythema with clear boundaries, different shapes and sizes, covered with multiple layers of silvery white scales, and Auspitz’s sign was positive. Admission diagnosis was a bone space occupying lesion in the left upper tibia which was thought to be a bone tumor, left knee joint degenerative changes, abdominal hernia after laparotomy,and psoriasis on both lower extremities.
Check after admission
On admission, a magnetic resonance imaging (MRI) of the left knee joint (Figure 2D-G)showed a wide range of abnormal mixed signals in the marrow cavities of the left femur, tibia, fibula and patella, the T1 weighted image and T2 weighted image (T2WI)bone marrow signal was decreased, and the spotted and patchy slightly longer T1 and T2 abnormal signals were scattered in the boundary, T2WI-fat suppression showed a slightly higher signal. In the upper lateral medullary cavity of the left tibia, an obscure border nodular lesion was observed. Its size in the sagittal position was about 2.6 cm ×1.9 cm, it showed long T1 and short T2 signals, its irregular annular edge had long T2 signals and the boundary was rough. Enhanced scanning of the lesion showed marginal patchy and mild irregular annular enhancement. No obvious absorption,destruction or periosteal reaction was observed in the adjacent cortical bones. No obvious abnormal signals were observed in the surrounding soft tissues. MRI findings were as follows: (1) A wide range of abnormal signals in the bone marrow cavity of the left femur, tibia, fibula and patella in the scanning area. Blood system diseases or metastases were considered; (2) Nodular lesions in the marrow cavity of the proximal lateral left tibia were likely to be osteogenic metastases; and (3) Left knee degenerative osteoarthrosis. Full-body bone imaging with 99mTc-MDP single photon emission computed tomography (SPECT) (Figure 2L) showed abnormal spotted radiated concentrations on the upper end of the left tibia, abnormal radiated symmetry concentrations in extremity joints, and an inflammatory lesion was considered. An MRI of the liver was performed (Figure 2A-C) which showed decreased liver diffuse signal, which was considered to be HC, spleen enlargement, gallstone and bilateral renal cyst. X-Ray of the skull, thoracolumbar spine, pelvis and chest (Figure 2H-K)showed thinned external and internal plates of the frontal bone and occipital bone,widened diploe, a high density stripy shadow in the low end of the occipital bone which was considered ligament calcification. The L4/5, L5/S1 intervertebral spaces were slightly narrow, lumbar vertebrae showed degenerative changes, vertebra proliferation, slightly rare bone trabecula and hyperosteogeny of bilateral acetabular.The high density shadow on the right upper abdomen was considered to be biliary calculi, density of the massive shadow increased in the right medioventral (range approximately 6.6 cm × 3.1 cm). No obvious abnormalities were noted in both lungs.
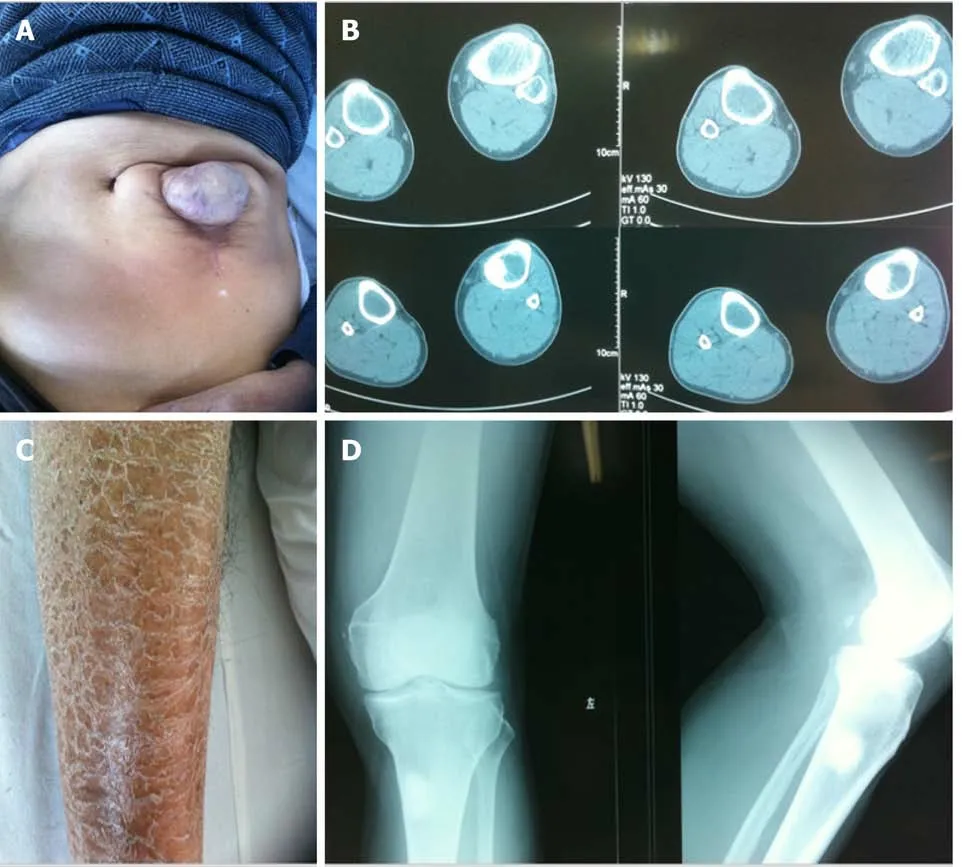
Figure 1 Clinical data of the patient. A: The patient did not heal after an exploratory laparotomy for acute abdomen at 40 years old and a ventral hernia was formed; B and D: Computed tomography and X-ray of the left knee joint showed a high-density shadow in the upper left tibia, which was considered to be an osteogenic nodule, the medullary cavity of the tibia was enlarged, and cortical bone was thinned; C: The patient suffered from psoriasis on both lower extremities.
A bone marrow smear (Figure 3A) showed that myeloproliferation was markedly active. Polychromatic erythrocytes and corpuscles were visible. Extracellular iron ++,intracellular iron -19%, +45%, ++28%, +++6%, and ringed sideroblasts of 2% were noted. A blood smear (Figure 3B) showed that mature erythrocytes differed in size,with an enlarged central light-stained area, and schistocytes (4%) and tear dropshaped erythrocytes (3%) were visible. In conclusion, the results of the bone marrow smear and peripheral blood smear showed obvious hyperplasia of the erythrocyte line,which was considered to be due to possible hemolytic anemia.
An acid hemolysis test, sucrose test, and heat hemolysis test were all negative.Autoantibodies including antinuclear antibody, anti-double-stranded antibody,etc.,were negative. Tumor markers including carbohydrate antigen (CA) 242, 724, 153, 125,carcinoembryonic antigen, alpha-fetoprotein, and prostate specific antigen were normal; low triiodothyronine syndrome was observed. No infection with hepatitis B virus was noted. Erythrocyte sedimentation rate was 4 mm/h and Bence-Jones protein was negative. Immune scatter turbidimetry showed that the hematuria κ light chain, λ light chain, and κ/λ levels were normal. Immunofixation electrophoresis showed that IgG, IgM, IgA, κ light chain, and λ light chain band were negative; serum protein electrophoresis bands were normal. The methemoglobin reduction test was decreased.Biochemical indices (Table 1) suggested that hemolytic anemia, microcytic hypochromic anemia, and erythrocyte distribution width (SD) were normal,erythrocyte distribution width (CV) was increased, and reticulocyte (Ret) count was increased. Iron metabolism was abnormal, manifested as iron overload characterizedby increased transferrin saturation (TS) and serum ferritin (SF).
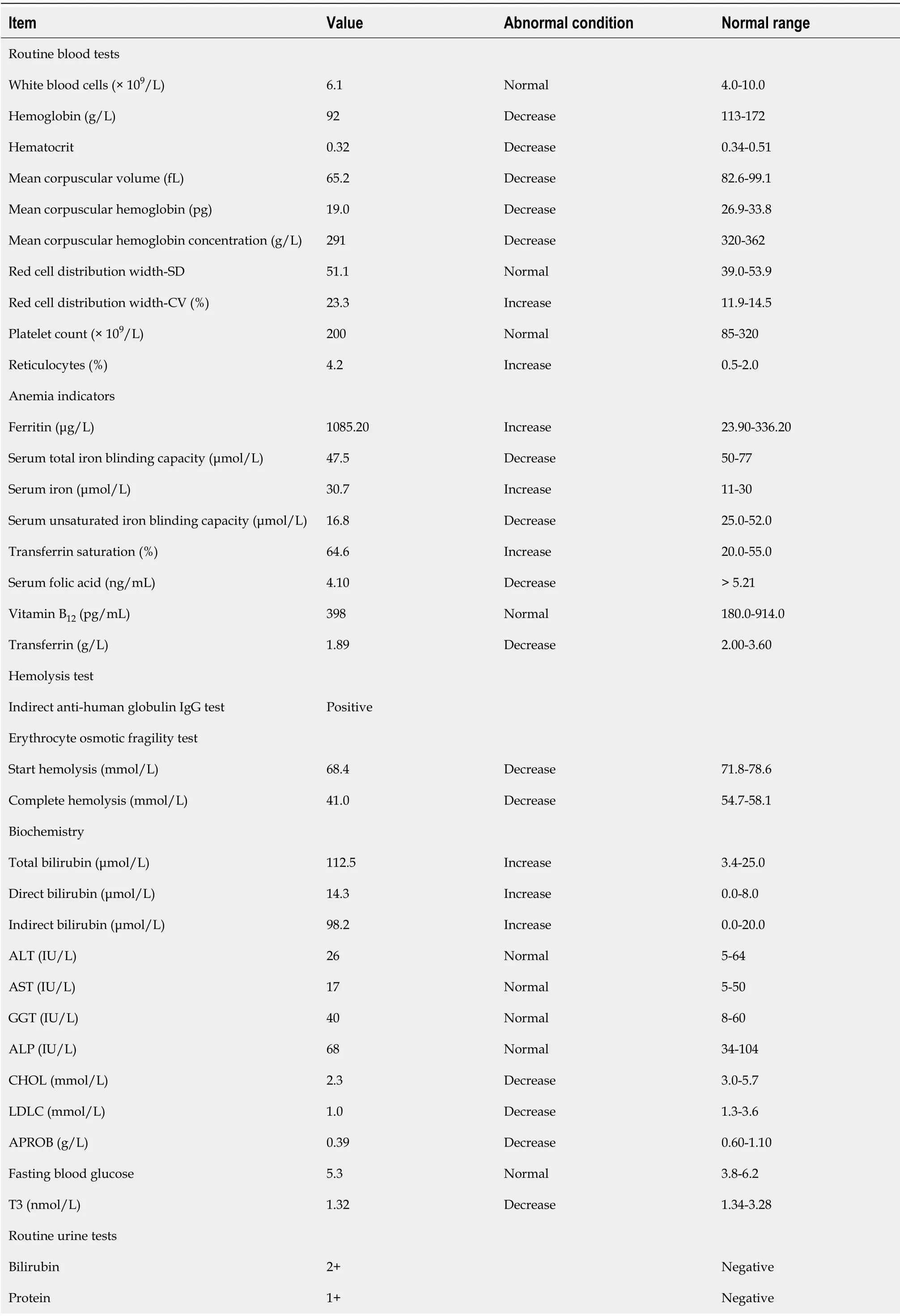
Table 1 Partial biochemical indicators in the patient with hemochromatosis

ALT: Alanine transaminase; AST: Aspartate transaminase; GGT: Glutamyltransferase; ALP: Alkaline phosphatase; CHOL: Total cholesterol; LDLC: Lowdensity lipoprotein cholesterol; APROB: Apolipoprotein B; T3: Triiodothyronine; RBC: Red blood cell.
On admission, according to the HC screening guide published by the American College of Physicians (ACP) in 2005, the indirect iron deposition index was as follows:Fasting TS for diagnosing the cut-off value of HH was female fasting TS > 50%, male fasting TS > 60%[9]. We considered this to be a HC patient and further detected the pathogenic gene to identify primary or secondary HC. In addition, the patient’s methemoglobin reduction rate decreased, which is an indirect reflection of G6PD activity, and as the incidence ofG6PDdeficiency in China is high, it was necessary to rule out this disease. This study was approved by the Ethics Committee of Fujian Provincial Hospital, No. K2015-031-01, and agreed by all patients.
RESULTS
Genetic diagnosis history
Ten years ago, the cost of whole exon sequencing was still high. With the informed consent of the patient and his family, we designed primers for PCR and Sanger sequencing of the HH common defect geneHFEandG6PDgene hot spot mutations.For example, the primer for the fragment of theHFEgene p.Cys282Tyr (C282Y)mutation (Figure 3D) was designed: L: 5’-TACCCCCAGAACATCACCAT-3’, R: 5’-GATCACAATGAGGGGCTGAT-3’, amplified product size 198 bp; the primer for theHFEgene p.His63Asp (H63D) mutation (Figure 3E): L: 5’-ACATGGTTAAGGCCTGTTGC-3’, R: 5’-ATGTGATCCCACCCTTTCAG-3’, amplified product size 245 bp. The primer for the fragment of theG6PDgene p.Arg459His (R459H, rs72554665)mutation (Figure 3G) was designed: R: 5’-CCTGCATACCTGTGGGCTAT-3’, L: 5’-AATATAGGGGATGGGCTTGG-3’, amplified product size 170 bp; the primer for theG6PDgene His32Arg (H32R, rs137852340) mutation (Figure 3H): L: 5’-TGCCTTGTTAACGAGCCTTT-3’, R: 5’-GATGCACCCATGATGATGAA-3’, amplified product size 157 bp. The annealing temperature of the above PCR was 55-58°C.Subsequently, we designed primers to detect theG6PDgene Val68Met (rs1050828).However, the results of several sites described above were all wild-type. Interestingly,an unaffected daughter of the proband showedHFEgene H63D heterozygous mutation (Figure 3F). H63D homozygotes have been considered to be less pathogenic in recent years. It has even been suggested that the addition of H63D genotyping does not greatly increase the efficiency of clinical evaluation, and adds confusion to the clinical interpretation of genetic testing results[10,11]. EASL clinical practice guidelines for HFE HC pointed out that for all patients with increased SF and TS that could not be explained by other reasons, the C282Y and H63D polymorphisms of the HC gene should be detected, and an elevated iron with C282Y homozygote could be the 1B evidence for the diagnosis of HH. C282Y/H63D heterozygote or H63D homozygote patients with increased SF (female > 200 μg/L, male > 300 μg/L), increased TS (female> 45%, male > 50%) or increased liver iron levels should first be investigated for other causes of hyperferritinemia as 1C evidence for the diagnosis of HH[12].
During the ten years of follow-up, the hot spot mutations ofG6PDandHFEgenes were repeatedly detected in the patient, and no significant mutation sites were found.The patient’s condition was stable and deferoxamine (DFO) was not used to treat iron overload.
With the decline in the price of all-exon sequencing scans in recent years, we used the Illumina HiSeq2500 sequencing platform to capture high-throughput sequencing of target regions two years ago. This time we screened genes related to HC, such asBMP2,FTH1,G6PD,HAMP,HFE,HFE2,SLC40A1,TFRand other 28 target genes. No suspected pathogenic or known pathogenicity of related gene mutation was detected.
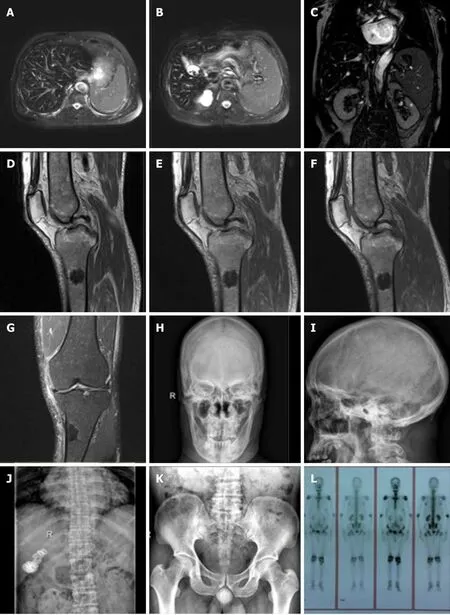
Figure 2 Imaging examination of the patient. A-C: Hepatic magnetic resonance imaging (MRI) showed a decreased hepatic diffuse signal, which was considered to be due to hemochromatosis, spleen enlargement, multiple gallstones and a renal cyst; D-G: MRI of the left knee joint showed wide abnormal mixed concentrations in the marrow cavity of the left femur, tibia, fibula and patella. Nodular lesions were noted in the marrow cavity of the proximal lateral left tibia. Left knee degenerative osteoarthrosis, degeneration and a tear in the anterior horn of the lateral knee joint meniscus, secondary degeneration in the medial meniscus anterior and posterior horn and the lateral meniscus posterior horn were also seen. A small amount of joint effusion was found in the left knee joint capsule, with soft tissue swelling around the knee joint; H-K: X-Ray showed thinned external and internal plates of the frontal bone and occipital bone, widened diploe, a high density striped shadow in the low end of the occipital bone which was considered ligament calcification. Hyperosteogeny of the vertebral body and bilateral acetabular, and slightly rare bone trabecula were observed. A high density shadow on the right upper abdomen which was considered biliary calculi, and increased density in a massive shadow in the right medioventral region were noted; L: The full-body single photon emission computed tomography showed an abnormal spotted radiated concentration on the upper end of the left tibia, abnormal radiated symmetry concentration in extremity joints, and an inflammatory lesion was considered.
In February 2019, we used the IlluminSNPa X10 and NovaSeq6000 platforms for PE150 sequencing (executed by iGeneTech) to detect single nucleotide polymorphisms covering all exons, small fragments of insertion and deletion, copy number variations,analyzed related gene exons of hereditary HC and secondary iron overload (such as iron-related anemia, hemolytic anemia, excessive iron overload caused by extraintestinal causes, chronic liver disease,etc.). Referring to the 1000G, ExAC, and gnomAD East Asian population databases, the mutations were screened at sites with a frequency of less than 0.01, with 415 mutations in the target capture region associated with HC and 377 mutations in the target capture region associated with psoriasis. We identified 8 mutation sites that are more likely to be consistent with the clinical phenotype: Including c.C4748T ofPIEZO1(NM_001142864), c.C2366T ofIFIH1(NM_022168), c.G1381A ofKRT8(NM_002273), c.A307G ofPOFUT1(NM_015352),c.C7764A ofFLG(NM_002016), c.A509G ofTGM5(NM_201631), c.303_304insGGC and c.300_301insGGCTTTGGAGGCGGC ofKRT2(NM_000423) (Table 2). Then performed PCR and Sanger sequencing. The primer for the fragment of thePIEZO1gene p.A1583V mutation (Figure 3I) was designed: R: 5’-CAAGGGAATGGCGAGGGTCG-3, L: 5’-CTGAGCACGGCGCAGTTC-3’, amplified product size 487 bp; The primer for thePOFUT1gene p.I103V mutation (Figure 3J): R: 5’-AATAAATGTCTAAAGTAGCCACGGG-3’, L: 5’-ATTTTGCCATGACATGTCTTGGG-3’, amplified product size 324 bp; The primer for theTGM5gene p.Q170R mutation (Figure 3K): R:5’-AGCGTTGGTCAGCAGTTCTAG-3’, L: 5’-GGGCAGTAAGGGAAAGGTGAC-3’,amplified product size 274 bp. The annealing temperature of the above PCR was 55-60°C.
From the sequencing results and clinical phenotypes, there was currently insufficient evidence to prove that these mutations were related to the hemolysis and iron overload characteristics of the patient. For example, thePIEZO1gene mutation may cause autosomal dominant dehydrated hereditary stomatocytosis (DHS).However, the phenotypes of DHS patients, such as blood smears, may have the characteristics of stomatocytes, macrocytic anemia,etc.The patient had microcytic anemia with no obvious characteristics of stomatocytosis, which does not seem to be consistent.
The final outcome of genetic diagnosis
In the following two months, we felt that we were not making any progress even with the use of high technology. Finally, considering that thalassemia is a common cause of hemolytic anemia in China, the genetic defects of this disease are common in the deletion or insertion of large fragment bases. The detection rate of this mutation is often the limitation of next-generation sequencing (NGS). Thus, does this patient we have been tracking for 10 years have thalassemia? We used the gap-PCR method to detect common mutations in the α-thalassemia gene (kit from Yaneng BIO) and simultaneously detected three globin gene deletion mutants (-α3.7, -α4.2, --SEA). These three types of mutations cover more than 95% of patients with α-thalassemia in the Chinese population, and finally found that the patient carried two deletion mutants -α4.2, --SEA(Figure 3C). These two mutants are common causes of α-thalassemia, and thalassemia causing HC is generally the result of severe β-thalassemia or long-term blood transfusion. However, such a serious non-long-term transfusion of secondary HC caused by -α4.2and --SEAmutation is rare. Does this mean that there is an“oligogenic disease” in which the remaining genetic mutations are involved in the onset of disease? Oligogenic diseases are polygenic genetic diseases that are caused by relatively few genetic effects, but some of them still have considerable effects. It can be seen that the presence of these trait-related mutations cannot be ignored when the mutation gene site is found by NGS.
DISCUSSION
Thalassemia is a group of hereditary small cell hemolytic anemias whose pathogenesis is a defect in the globin gene, such that one or more of the globin α, β, γ, δ peptide chains in hemoglobin are reduced or unable to synthesize (of which β and α peptides chain mutations are common), leading to changes in the composition of hemoglobin.The clinical symptoms in this group of diseases vary in severity, mostly manifested as chronic progressive hemolytic anemia. The current new classification of thalassemia disease is based on the patient’s clinical severity and is simplified into transfusiondependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT),which also includes all other forms of thalassemia syndrome, such as α-thalassemia,hemoglobin E/β-thalassemia and combined α- and β-thalassemia[13-15]. Patients with αthalassemia (OMIM#604131) are mainly distributed in tropical and subtropical regions, and are more common in southern China. The disease is divided into two types, the deletion type and the non-deletion type. The deletion type of thalassemia includes α+thalassemia (Southeast Asian type, --/αα, --SEA) which lacks two α genes on chromosome 16 and α+thalassemia (-α/αα) which lacks an α gene. The α+thalassemia usually has -α3.7, -α4.2deletions. The common deletion type in α-thalassemia in China is: -/αα, -α3.7α/αα and -α4.2α/αα. Different types of α-thalassemia patients have different numbers of α-globin genes deleted in the body, and the more α genes deleted, the more serious the disease is[16,17]. The deletion type in this case of αthalassemia is also a relatively common type of Southeast Asian mutation, while secondary HC with a severe osteoarthrosis phenotype caused by NTDT is rare.Therefore, we speculate that there may be some minor-effect iron overload-related gene mutations involved in iron homeostasis imbalance. We noted a rare missense mutation on the piezo typePIEZO1gene exon 35, which was co-located on chromosome 16 with the globin gene: c.C4748T (p.A1583V), with a very low minor allele frequency (MAF) and moderately harmful protein function prediction.Mutations in thePIEZO1ion channel gene (PIEZO1, 611184.0001) can cause phenotypes of DHS with or without pseudohyperkalemia and/or perinatal edema, the phenotype includes anemia, chronic hemolysis, jaundice, serosal effusion, thrombosis,hepatosplenomegaly, cholelithiasis, hemoglobinuria, fatigue,etc.Osmotic gradient ektacytometry is a useful characteristic diagnosis method for this disease[18]. Therefore,we speculated that the p.A1583V mutation of thePIEZO1gene might participate in the α0and α+mutations of the globin α gene and aggravate the degree of iron deposition,which caused a cumulative damage effect and was similar to oligogenic disease. The DHS caused by thePIEZO1mutation belongs to one of the red blood cell (RBC)membrane disorders as well as hereditary spherocytosis (HS) and hereditary elliptocytosis (HE). For example, HS is a group of genetically heterogeneous anemias with a broad spectrum. The clinical phenotypes vary in severity and can range from asymptomatic to severe blood transfusion-dependent forms. Different degrees of clinical manifestations exist in the same family. This intraclass genetic heterogeneity can be attributed to the co-inheritance of genetic variations involving multiple erythrocyte defects, such as thalassemia, enzymopathies and Gilbert syndrome[19]. Did this secondary HC we found, show a distinct HC osteoarthritis phenotype due to this similar co-inheritance phenomenon of thalassemia and DHS? Or didPIEZO1gene mutation p.A1583V play a minor-effect auxiliary pathogenic role?
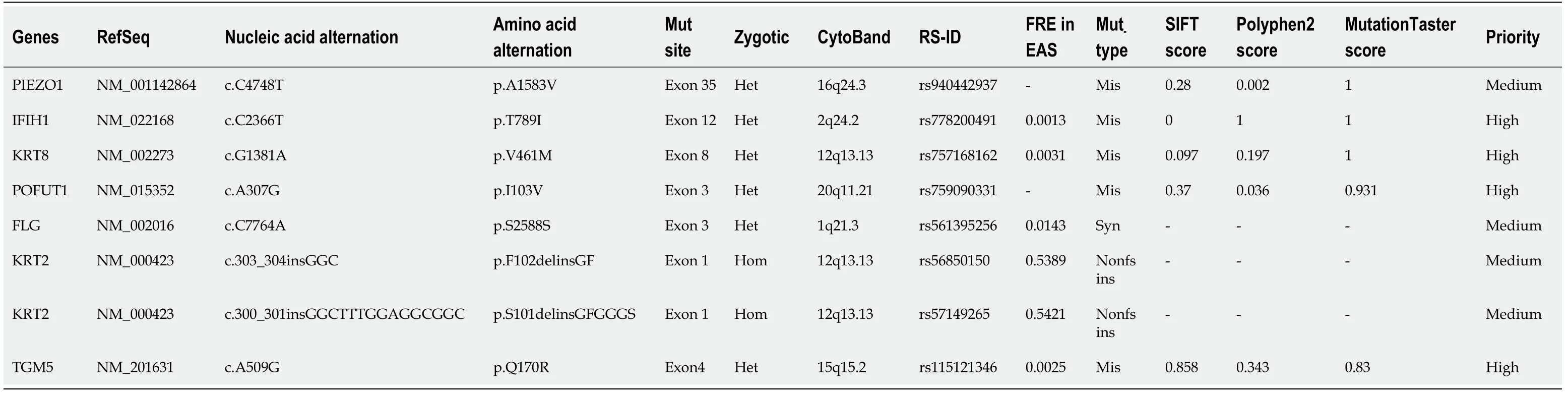
Table 2 Possible mutation sites in the patient associated with hemochromatosis or psoriasis
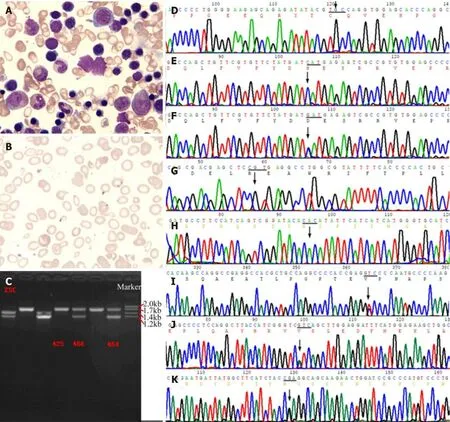
Figure 3 Bone marrow smear and genetic analysis. A: Bone marrow smear showed erythroid cell hyperplasia, with polychromatic normoblasts.Nucleocytoplasmic development was imbalanced, with visible binuclear and mitotic types. Mature erythrocytes differed in size, which included partially large spherocytes 3%, teardrop 4%, irregular 5%, and fragmented 3% cells. Visible polychromatic erythrocytes and corpuscles were observed. Extracellular iron ++,intracellular iron -19%, +45%, ++28%, +++6%, ringed sideroblasts 2%; B: Blood smear showed obvious erythrocyte hyperplasia, possibly due to hemolytic anemia; C:Two heterozygous deletion mutants of the -α4.2, --SEA carrying globin gene were finally detected; D-H: The patient had p.C282Y and p.H63D of HFE gene, p.R459H and p.H32R of G6PD gene which were wild-type; F: His daughter carried a heterozygous p.H63D mutation of HFE gene; I-K: The patient showed p.A1583V mutation of PIEZO1 gene, p.I103V mutation of POFUT1 gene, and p.Q170R mutation of TGM5 gene.
The genetic background of RBC membrane disorders is a mutation in the gene encoding membrane or cytoskeletal proteins, transmembrane transporters or channels,resulting in membrane structural defects and membrane permeability changes. It is very important in all cells based on the spectrin membrane skeleton structure.Membrane skeleton structure defects are the cause of multiple hemolytic anemia[20].The mechanoreceptorPIEZO1is the largest ion channel ever discovered and a fundamental regulator of RBC volume homeostasis, which regulates the mechanotransductive release of ATP in human RBCs[21]. In the functional studies of several families, the missense mutation ofPIEZO1has been proved to be the cause of DHS or xerocytosis, which delays the inactivation of the erythrocyte dehydration channel[18,22].
Obviously, hereditary anemia such as RBC membrane disorders or hemoglobinopathy is highly genetically heterogeneous and is a genetically deficient disease with a highly overlapping clinical phenotype. Under certain conditions, except for some of the obvious phenotypic variations which can be explained by a high degree of genetic heterogeneity, it is often difficult and complicated to distinguish the disease type from the patient’s clinical manifestations. Sometimes the carrier of the mutation does not show any symptoms, so it is easily ignored. Therefore, it is necessary to observe the morphology of peripheral blood smears, and to understand the RBC function by examining the RBC osmotic fragility (OF) test, ektacytometry,etc., and to conduct an in-depth analysis of the genetic pattern of the family. Genetic analysis of hereditary hemolytic disease of this fuzzy phenotype is particularly important[20]. However, due to the different formation of mutations, the sensitivity of DNA detection methods is different, so the choice of DNA detection methods becomes crucial. For example, karyotyping analysis is applied to chromosome number abnormalities or chromosomal structural abnormalities of large fragments above 5 Mb;fluorescent in situ hybridization assists in the diagnosis of chromosomal diseases,determining the source, location and number of abnormal chromosomes;chromosomal microarray analysis can detect the copy number variation above 100 kb;multiplex ligation-dependent probe amplification is mainly used for detection of target fragment gene copy number changes (such as DMD, SMA, 5p deletion, 22q deletion);NGS includes panel, clinical exome sequencing, whole exome sequencing, and whole genome sequencing, but NGS still lacks sensitivity to large copy number changes.Sanger sequencing is often used as a detection method for single genetic diseases with accurate pathogenic genes or pathogenic sites or as a verification technique for NGS results. In this patient, we finally detected the alpha globin gene mutation by gap-PCR.At present, gap-PCR is still the best solution for screening the three deletion mutants (-α3.7, -α4.2, --SEA) of α-thalassemia patients in the Chinese population. In fact, we are looking forward to the emergence of a more comprehensive and low-cost new genome sequencing technology to make it easier to find the disease-causing genes in highthroughput.
Interestingly, this patient also has a hereditary disease: Psoriasis in both lower extremities. Psoriasis is a chronic inflammatory skin disease characterized by hyperproliferation of the epidermis and vascular remodeling as the main clinical phenotype. The pathogenesis is currently unknown, and may be related to geneticenvironment and its gene-gene interaction (also known as epistatic effects). It is a complex polygenic disease. The gene interaction refers to the interaction between nonalleles, and the interaction of different genes that affects the characteristic expression of disease phenotypes. More and more studies have found that it is an important pathogenesis of complex diseases[23]. In recent years, genome-wide association studies of complex diseases have found that more than 40 susceptibility genes are associated with the pathogenesis of psoriasis[24]. We found that this case had some mutation sites with a very low frequency of MAF related to skin traits such as:IFIH1(p.T789I),KRT8(p.V461M),POFUT1(p.I103V),FLG(p.S2588S),KRT2(p.F102delinsGF and p.S101delinsGFGGGS),TGM5(p.Q170R). Among them, IFIH1 (p.T789I) protein function prediction is highly harmful, and we speculated that there may be an interaction between these mutation sites, thus participating in the chronic inflammation of psoriasis.
The severity of the clinical phenotype profile of thalassemia depends on the severity of mutations in the globin gene and the common inheritance of other important genes.A characteristic clinical phenotype of this case is that the abnormal iron deposition in the liver, spleen and whole body are more serious. The incidence of hemochromatosis osteoarthropathy is 43%-57%, of which only 55% have joint pain symptoms. X-ray can identify bone and joint lesions, such as joint cystic changes and edge hardening changes, which are common in the second and third metacarpophalangeal joints, joints of the knees, hips, and ankles can also be affected, which can be the first or only manifestation[25]. Acute abdomen which was noted 30 years ago may be abdominal pain caused by iron deposition in viscera. After an exploratory laparotomy, the wound was difficult to heal and formed an abdominal hernia, which may be attributed to the decreased anti-infective ability of the tissue and lack of tissue repair ability caused by serious iron deposition.
The main mechanism driving the iron loading process is the continued increase in iron deposition after TDT transfusion therapy, whereas NTDT involves secondary intestinal absorption of iron due to ineffective erythropoiesis and excessive inhibition of hepcidin. Due to different iron loading mechanisms and iron accumulation rates,iron overload has different effects on different organs, and the SF levels and imaging are also different. In TDT, infused RBCs are phagocytosed by macrophages in the reticuloendothelial system after cell senescence. Then unstable cellular iron is released into plasma to bind transferrin. When bound transferrin is saturated, non-transferrinbound iron readily enters the visceral or other tissues through the calcium channel.The resulting reactive oxygen species cause organ function decline, apoptosis or necrosis[26,27]. The main complication of TDT iron overload is cardiac siderosis,arrhythmia and heart failure secondary to cardiac siderosis are the main causes of death. Other complications include chronic hemolysis, chronic viral hepatitis, liver fibrosis, endocrine diseases such as bone disease, gonad hypofunction,hypothyroidism, hypoparathyroidism, and diabetes[28]. However, The main organ affected by iron overload in NTDT is the liver rather than the myocardium[29].Common complications include osteoporosis, extramedullary hematopoiesis,osteoarthrosis, hypogonadism, liver disease, renal insufficiency, thrombosis,pulmonary hypertension, leg ulcers,etc.Compared with TDT, hypothyroidism, heart failure and diabetes are less common in NTDT. Besides, the prevalence of iron overload in thalassemia varies from region to region. For example, more than 25% of patients with β-thalassemia major in Southeast Asia have cardiac siderosis complications, compared with 15%-20% in Europe and the Middle East[30]. In addition,the complications of TDT and NTDT also occur at different times. For example,patients with TDT have an early iron overload phenotype after 10 to 20 transfusions.NTDT patients often have iron overload after the age of 10 to 15[26,27,31]. The difference in these complications may be attributed to the fact that iron overload or the rate of iron deposition is slower in NTDT, but the mechanism is still unclear. In terms of treatment, iron chelators with different therapeutic effects were selected based on different types of iron loads occurring in different organs: DFO, deferiprone and deferasirox[27].
CONCLUSION
Despite advanced genetic testing technology, the selection of genetic detection methods for HC still needs to be based on an in-depth study of the clinical manifestations of the disease.
ARTICLE HIGHLIGHTS
Research background
Hemochromatosis (HC) is an iron overload disease caused by iron metabolism disorders. It can be divided into two major categories of primary and secondary in terms of etiology. Different genetic heterogeneity and different gene mutations in HC increase the difficulty in identifying genetic causes and diagnosis.
Research motivation
This complex clinical phenotype is characterized by a difficult case of HC including liver and bone tissue damage. It had been misdiagnosed as a metastatic tumor, and the genetic cause was not clear.
Research objectives
To investigate the genetic background of this patient with HC complicated by psoriasis on both lower extremities.
Research methods
The patient underwent detection of several hot spot mutations of theHFEandG6PDgenes by next-generation sequencing, but no responsible gene mutation was found.Finally, the thalassemia gene was detected by gap-PCR.
Research results
The patient was found to carry the -α4.2and --SEAdeletion mutants of the globin gene.These two types are common causes of Southeast Asian α-thalassemia, but rarely cause severe widespread non-transfusion secondary hemochromatosis osteoarthropathy. Thus, the simultaneous presence of the auxiliary superposition effect of the rare missense mutation ofPIEZO1gene (NM_001142864, c.C4748T,p.A1583V) was considered.
Research conclusions
The selection of genetic detection methods for HC still needs to be based on an indepth study of the clinical manifestations of the disease.
Research perspectives
Due to the different mutation forms and the different sensitivity of DNA detection methods, the selection of DNA detection methods is very important. We are looking forward to the emergence of a more comprehensive and low-cost new genome sequencing technology, so that it is easier to identify HC pathogenic genes with high throughput.
ACKNOWLEDGEMENTS
The authors also would like to thank the patient and his family for their contribution to this study.
 World Journal of Clinical Cases2020年23期
World Journal of Clinical Cases2020年23期
- World Journal of Clinical Cases的其它文章
- Understanding the immunopathogenesis of COVID-19: Its implication for therapeutic strategy
- What is the gut feeling telling us about physical activity in colorectal carcinogenesis?
- Latest developments in chronic intestinal pseudo-obstruction
- Correlation between ductus venosus spectrum and right ventricular diastolic function in isolated single-umbilical-artery foetus and normal foetus in third trimester
- Clinical efficacy of integral theory–guided laparoscopic integral pelvic floor/ligament repair in the treatment of internal rectal prolapse in females
- Treatment of Kümmell’s disease with sequential infusion of bone cement: A retrospective study