
可变性红斑角皮症1例
2020-03-26 01:34张琪李超刘家蕊钟维朱磊黄钰淇李青雯陈永锋
皮肤性病诊疗学杂志 2020年1期
张琪, 李超, 刘家蕊, 钟维, 朱磊, 黄钰淇, 李青雯, 陈永锋
(1.广东医科大学,广东 湛江 524023;2.南方医科大学皮肤病医院,广东 广州 510091;3.深圳市慢性病防治中心,广东 深圳 518020)
1 临床资料
患儿男,2岁零3个月,因全身多处红斑、脱屑伴瘙痒10月余就诊。10月余前,患儿无明显诱因下双肘窝出现红斑伴脱屑,未予诊治,皮疹逐渐累及颈部、腋下、四肢伸侧及腰背部,并伴有轻度瘙痒。皮疹受热后可稍有加重,部分皮疹可自行消退,但仍有新皮损不断出现。患儿曾就诊于外院,考虑“剥脱性皮炎”,予外用药物(具体用药不详)治疗后效果欠佳。既往体健,父母非近亲结婚,否认家族中有类似疾病史。
体格检查:各系统检查未见异常。皮肤专科检查(图1A、1B):颈部、双腋下、脐周、四肢可见对称分布、大小不等的片状、环状、地图状红色、褐色角化性斑片及斑块,边界尚清,表面粗糙,上覆薄层糠状鳞屑,Auspitz征阴性,其间散在分布绿豆至黄豆大小角化过度性丘疹、斑丘疹,口腔粘膜、肛门生殖器、毛发、牙齿及指(趾)甲未见异常。
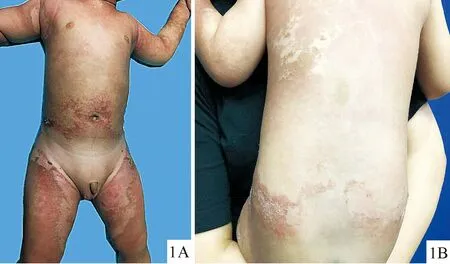
图1可变性红斑角皮症患儿躯干(1A)、背部(1B)可见角化性、鳞屑性红褐色斑片、斑块,边界尚清
Fig.1Clinical pictures of the patient with erythrokeratodermia variabilis: demarcated hyperkeratotic, scaly erythematous on the trunk (1A) and the back (1B).

图2可变性红斑角皮症患儿腹部皮损组织病理: 表皮角化过度伴角化不全,颗粒层存在,棘层不规则肥厚,轻度乳头瘤样增生。表皮突不规则向下延伸,真皮浅层毛细血管增生、扩张,少量淋巴细胞浸润(HE,2A:40×;2B:100×)
Fig.2Histopathological pictures of the abdomen skin lesion: hyperkeratosis,focal parakeratosis, spinous acathosis, mild papilloma-like hyperplasia,trochanterellus, downward extension of rete ridges, proliferation and dilation of capillaries with lymphocytic infiltrate in the superficial dermis(HE staining,2A:40×;2B:100×).
实验室检查:微量元素六项(锌、铁、铜、镁、钙、铅)正常。
取腹部红斑处皮损组织行病理检查示(图2A、2B):表皮角化过度伴角化不全,颗粒层存在,棘层不规则肥厚,轻度乳头瘤样增生。表皮突不规则向下延伸,真皮浅层毛细血管增生、扩张,血管周围有稀疏的淋巴细胞浸润。
基因检测:患儿外周血全外显子基因测序提示:GJB4基因杂合突变(图3)。
诊断:可变性红斑角皮症。
治疗:予口服复方氨肽素片2片/次,每天2次,盐酸西替利嗪滴剂2.5 mg/次,每天1次,辅以尿囊素乳膏及氢化可的松乳膏外涂治疗。1个月后电话随访,瘙痒症状明显好转,皮疹稍有好转,3个月后患儿皮疹基本消退(图4A、4B)。

图3 可变性红斑角皮症患儿外周血GJB4基因测序图(部分)Fig.3 The gene sequencing map of GJB4 in the case with EKV (part).
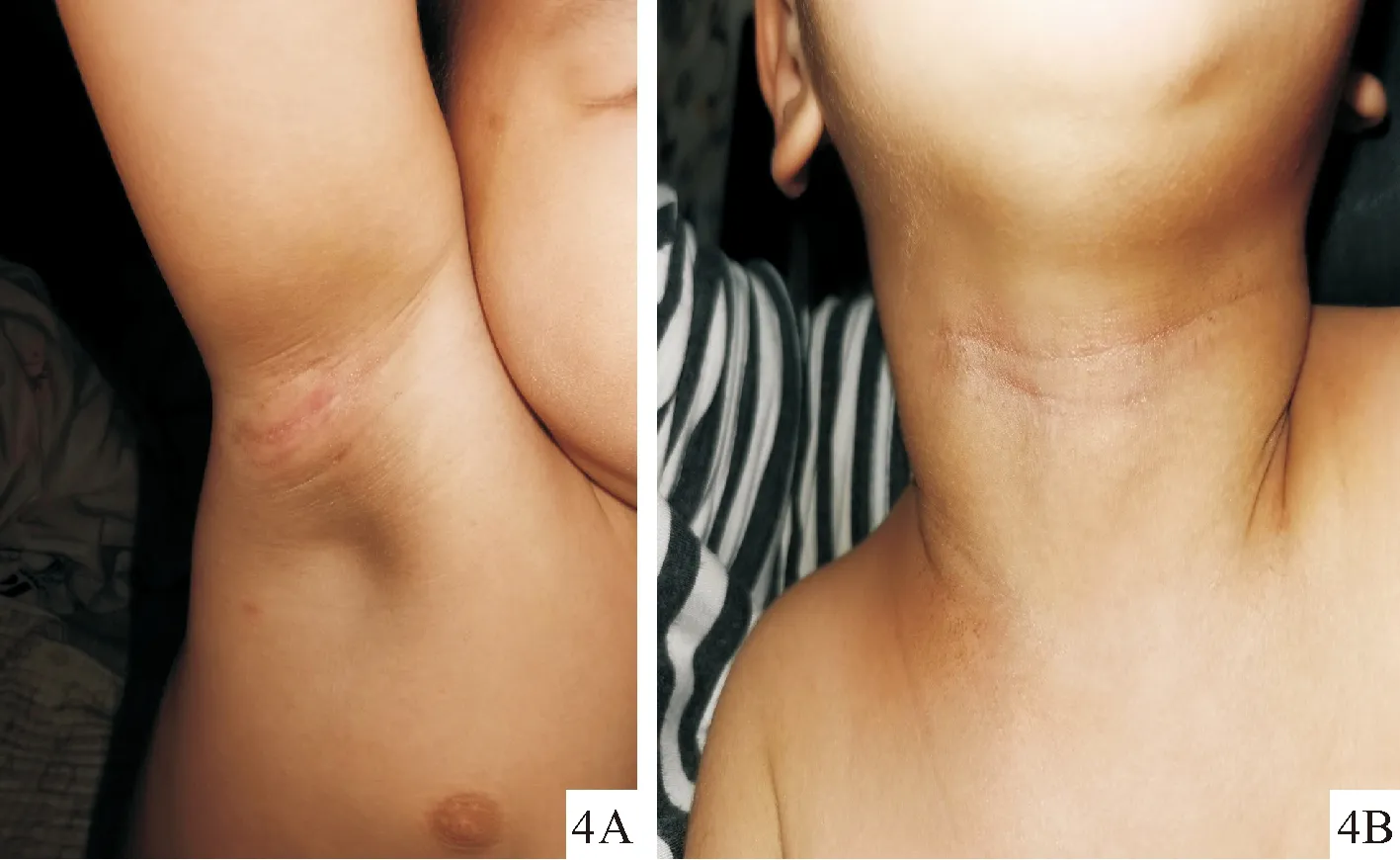
图4可变性红斑角皮症患儿(4A、4B)治疗3个月后红斑鳞屑基本消退
Fig.4The erythematous and scales subsided (4A,4B) after3months of treatment.
2 讨论
可变性红斑角皮症(erythrokeratodermia variabilis, EKV),又名可变性图形红斑角化症。于1925年由Mendes da Costa命名,故又名Mendes da Costa病[1]。本病是一种少见的、被认为与遗传相关的原发性皮肤角化病,可能为常染色体不规则显性遗传,也有常染色体隐性遗传的报道,参与发病的基因主要为编码缝隙连接蛋白GJB3或GJB4基因突变所致[2]。
本病常为幼年起病,患儿出生不久至3岁内可发病。特征性表现为边界清楚的红斑及角化过度性斑片,通常有2种临床表现:一种为对称分布、形态多样的红斑,红斑大小、数量、位置可因情绪、气温变化而在数周内或数日内,甚至数小时内发生明显变化;另一种常在正常皮肤上出现红色、紫红色、棕黄色角化性斑片或斑块,边界清楚,形状不规则,有些状如地图,常无自觉症状,偶有轻度瘙痒[3]。皮损可发生在任何部位,但多见于四肢伸侧及腋下、臀部,通常为夏重冬轻。本病终身不愈,但一般不影响健康。本病组织病理学改变无特异性,常表现为棘层肥厚和乳头瘤样增生[3]。
本例患儿于1岁零5个月时无明显诱因起病,临床表现为全身多处对称性、边界清楚的角化性红斑、暗红斑,表面粗糙,上覆糠状鳞屑,自觉症状轻。查血微量元素六项正常,全血基因外显子测序提示GJB4基因杂合突变,组织病理学改变无特异性。诊断明确,予口服复方氨肽素片、盐酸西替利嗪滴剂,外涂激素乳膏及保湿乳膏有效。
本病主要需与先天性大疱性鱼鳞病样红皮病(congenital bullous ichthyosiform erythroderma,CBIE)等相鉴别。CBIE是一种常染色体显性遗传病,是由角蛋白1 (KPT1)和角蛋白10 (KPT10)的基因杂合变异所致[4]。大部分患儿出生时即可起病,临床上主要表现为反复发作大疱、糜烂、红斑、结痂伴脱屑,症状随着年龄增长而减轻。虽然EV与CBIE有一些共同的临床表现和皮肤病理特征,但两者可以通过基因检测来鉴别诊断。
目前本病无特效疗法,可予口服异维A酸或阿维A,辅助以外用糖皮质激素软膏及保湿霜、尿素霜。有学者认为维A酸类药物联合补骨脂素长波紫外线照射有较好的疗效。抗组胺药对瘙痒性皮损有效[1,5]。但停药后可复发[6]。
综上所述,有相当一部分皮肤病发病机制与遗传相关,诊疗过程中须做好专科查体及病史询问,必要时行组织病理检查甚至基因筛查。对于该类与基因突变相关的疾病,皮损治愈几率低,需考虑患者个体差异而制定不同的、长期的指导治疗,并定期随访。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
现代仪器与医疗(2022年1期)2022-04-19
中国美容医学(2022年2期)2022-03-17
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
皮肤病与性病(2021年3期)2021-07-30
皮肤病与性病(2021年3期)2021-07-30
家庭医药(2018年2期)2018-02-09
东方教育(2017年14期)2017-09-25
家庭医药(2016年4期)2016-05-04
