
The Janus face of N-terminal lysines in α-synuclein
2020-03-25 10:22AnaBelénUceda,LauraMari?o,MiquelAdrover
中国神经再生研究(英文版) 2020年10期
Parkinson’s disease (PD) is the second most prevalent progressive neurodegenerative disorder after Alzheimer’s disease. PD usually starts with a tremor in the extremities (usually in the hands) and gradually evolves with other symptoms such as bradykinesia, muscle stiffness, impaired posture, loss of automatic movements or speech changes. These symptoms worsen as the condition progresses and eventually lead to death.
PD results from deterioration of dopamine-producing neurons in substantia nigra, which are especially vulnerable owing to their extensive branching and abnormally large number of synapses. Post-mortem studies of patients dying from PD have shown them to share a characteristic trait, namely: accumulation of intraneuronal protein deposits known as “Lewy bodies (LBs)”. LBs also cause dementia with LB (DLB),a condition that shares the symptoms of PDs albeit in a different sequence. Thus, dementia and memory loss always appear early in DLB but years after diagnosis in PD. In contrast, movement changes appear early in PD but not always occur in DLB. These differences have been ascribed to the increased amyloid load in the striatum, claustrum, cortex and globus pallidus in DLB relative to PD. In any case, LBs interfere with neuronal trafficking, disrupt membranes and sequester proteins.
The main component of LBs is α-synuclein (αS), a small monomeric protein that is highly abundant in the presynaptic terminal of dopaminergic neurons. Although αS has been associated with a large number of neuronal functions, its precise biological role remains unknown. For instance, αS is involved in the regulation of the presynaptic vesicular pool and in the protection of nerve terminals against injury. Also, it acts as a chaperon for synaptic SNARE proteins, regulates neuronal redox balance, inhibits apoptosis, helps regulate glucose levels and modulates calmodulin activity, among other functions.
Based on its physicochemical properties, αS is a highly aggregation-prone protein. Thus, its native form tends to aggregate through a nucleation—polymerization mechanism whose rate-determining step is the formation of nuclei that subsequently grow through monomer association to a size where incorporation of additional monomers is no longer favourable. The early stages of αS aggregation are populated by oligomeric and protofibrillar assemblies, which have been deemed the most neurotoxic species in the aggregation pathway. These oligomers bind to neuronal membranes and rouse the formation of reactive oxygen species, thus facilitating PD. Subsequently, these preliminary assemblies further evolve into mature amyloid fibrils consisting of two protofilaments that intertwine to form a left-handed helix (Li et al.,2018). The V37-Q99 stretch of each protofilament in a fibril adopts a Greek-key β-sheet motif that is not present in the oligomers formed at the early stages of aggregation (Carija et al., 2019); therefore, it cannot be used as a template to design PD therapies. Finally, αS fibrils clump into LBs.
αS aggregation is triggered by a number of factors including increased expression of αS encoding gene (SCNA) by effect of duplication or triplication. Genetic mutations such asA53T,A30P,E46KorG51Dcan also promote αS aggregation and lead to early-onset forms of PD.Complexation of αS by metals increases its aggregation rate and causes the formation of neuronally toxic reactive oxygen species. Because αS in LBs usually exhibits post-translational modifications (PTMs), part of scientific research in this area has focused on unveiling the precise effect of the PTMs on the aggregation propensity of αS. αS isolated from LBs has been found acetylated, phosphorylated, ubiquitinated, nitrated,sumoylated, truncated, oxidized and glycosylated (Chen et al., 2019).Nα-acetylation causes no aggregation of αS, whereas phosphorylation of S129 promotes it, while that occurring on Y39, S87, Y125 and Y133 inhibits it (Chen et al., 2019). Although ubiquitination and sumoylation of Lys have site-dependent effects, they usually delay or even inhibit αS aggregation (Chen et al., 2019). Tyr nitration on Y39, Y125, Y133 and/or Y136 stabilizes αS oligomers but inhibits fibril formation. C-terminal truncated αS exhibits a higher aggregation propensity than does fulllength αS, but truncation ofN-terminal Lys inhibit aggregation (Chen et al., 2019). Finally, Met oxidation substantially stabilizes neurotoxic αS oligomers (Glaser et al., 2005), whereasO-GlcNAcylation completely inhibits aggregation (Levine et al., 2019).
Precisely understanding the physiological and pathological roles of αS requires a profound analysis of its sequential and structural features,and also of the changes induced by its associated mutations and PTMs.The primary sequence of αS comprises three different domains, namely:(a) anN-terminal lipid-binding domain (M1-K60) containing an imperfect conserved repeat KTKGEV that acts as the membrane anchor region of αS; (b) a non-amyloid-β central domain (NAC; E61-V95)with a highly hydrophobic motif that is responsible for αS aggregation through a conformational change from a random coil conformation to the β-sheet structure found in mature amyloid fibrils (Li et al., 2018);and (c) a C-terminal acidic domain (K96-A140) that is required by αS to interact with its biological partners, and is highly rich in acidic and disorder-promoting residues counteracting the tendency of the NAC domain to aggregate (Figure 1A).
The monomeric form of native αS in neuronal cytosol adopts an intrinsically disordered structure; however, its weighted average conformation is not fully extended as it natively exhibits interactions between the NAC and C-terminal domains, and also between theN-terminal and C-terminal domains. When these long-range interactions are perturbed (e.g., as a result of mutations, pH changes or metal chelation),the NAC domain is exposed and aggregation triggered. However, the presence of lipid membranes, droplets, rafts or vesicles causes theN-terminal and NAC domains to adopt an α-helical structure that mediates binding of αS to membranes (Chandra et al., 2003).
αS can adopt either a broken or an extended α-helix conformation depending on the curvature of the membrane. When αS binds to membranes with a large diameter (i.e., large unilamellar vesicles), it adopts an elongated α-helical structure; on the other hand, when it binds to small, highly curved vesicles, it acquires a broken α-helix conformation(Figure 1B). The membrane binding process occurs through a cooperative effect of the different KTKGEV repeats and requires the lipids to contain acidic head groups. This suggests a direct interaction of cationic Lys in theN-terminal and NAC domains with polar heads in the lipids. In fact, truncation of theN-terminal domain has been found to drastically reduce αS-lipid binding (Vamvaca et al., 2009).
The primary sequence of αS includes an abnormally large number of Lys residues (15) that are located mainly in theN-terminal domain.These Lys residues account for 10.7% of the entire sequence (Figure 1A) and are fully conserved among species, which suggests a crucial biological role in αS. As state above, Lys residues in αS are essential for its binding to membranes. Because the residues fall normal to the helical axis, they facilitate interaction with phosphate head groups in lipids.
Neuronal αS clearance is one other very important process because it prevents accumulation of αS aggregates. The fact that a variety of Lys residues including K6, K10, K12, K21, K23, K32, K34, K43, K46 and K96 exhibit ubiquitination suggests that the clearance is effected by the ubiquitin—proteasome system (Chen et al., 2019). The process may arise from the action of various E3 ubiquitin ligases but also by the deubiquitinase USP9X, which regulates ubiquitinated αS levels. Lys might also be crucial for direct proteolysis of extracellular αS during interneuronal transmission of αS oligomers. In fact, some extracellular proteases can degrade αS by cleavage at specific points. Thus, plasmin and the serine protease KLK6 can cleave αS at S9 and S42, which are located near K10 and K43, respectively. In addition, matrix metalloproteinase 3 cleaves αS at sites near K58, K80, K96 and K102. Lys residues in αS have also been found in sumoylated form, which is more soluble and less prone to aggregate (Abeywardana and Pratt, 2015). Also, sumoylation of K96 and K102 has been found to direct αS to extracellular vesicles.
Although Lys residues seem to be crucial for αS metabolism and biological function, they may harbour a dark side. Thus, the high nucleophilicity of their side chain can make the residues perfect targets for the catabolic aldehydes that occur at considerably increased levels in Parkinsonian brains. Lys changes may even be promoted by the high concentration of αS in neurons (~40 μM) and also by their unshielded side chains—a typical feature of disordered proteins. Hence, the physiological role of αS might be hampered by non-enzymatic post-translational changes in its Lys residues. The effect of Lys changes on αS function has scarcely been studied to date. Consequently, and given the increasing prevalence of PD, this topic demands urgent attention from the scientific community. Also, our hypothesis is reinforced by the fact that αS isolated fromin vivosamples contains Lys residues that are modified by a broad set of PTMs. The associated changes arise largely from the reactions of αS with (a) 3,4-hydroxyphenylacetaldehyde, one of the main products of dopamine metabolism; (b) 4-hydroxynonenal,which forms by lipid peroxidation; and (c) methyglyoxal, a prominent product of intraneuronal glycolysis (Figure 1C). The potential effects of these aldehydes on αS are discussed from different point of views below.
Lys modification seemingly increases αS toxicity. For instance, LBs of people simultaneously having PD and diabetes mellitus contain abnormally high concentrations of advanced glycation end-products(AGEs) but especially MOLD andNε-(carboxyethyl)lysine (CEL). Both AGEs arise from the action of methyglyoxal, and facilitate aggregation of αS and accumulation of toxic oligomers. Extracellular glycated αS might also interact in a direct manner with the transmembrane receptor for advanced glycation end-products to activate pro-inflammatory genes. Altogether, this might explain the increased prevalence of PD in diabetic patients (~38%; Yue et al., 2016). Also, covalent modification of Lys by 3,4-hydroxyphenylacetaldehyde favors the formation of toxic oligomers (Follmer et al., 2015), whereas the reaction of αS with 4-hydroxynonenal inhibits fibrillization while favoring the formation of soluble off-pathway oligomers (Qin et al., 2007).
Lys modification might affect the vesicle binding ability of αS. The chemical modification of Lys residues may directly deplete their positive charges and alter the electrostatic potential of αS (especially in theN-terminal domain) as a result (Figure 1D). Without positive charges,it is nearly impossible for αS to interact with lipid vesicles, so equilibrium is shifted to the unbound form (Figure 1B), thereby altering neurotransmitter release and assembly of the SNARE complex, which are the two most important functions ascribed to the αS-lipid complex.
Aldehydes can compete with the naturally occurring post-translational modifications. Chemically modified Lys residues in αS cannot be ubiquitinated or sumoylated. This may interfere with αS clearance and intraneuronal accumulation of the protein; also, it may modify the interaction pattern of αS with other proteins—and hence, cell signalling. Moreover, Lys modifications may hinder direct proteolysis of extracellular αS through steric hindrance to cleavage. For instance,modifications on K58, K80, K96 and K102 may significantly diminish the extracellular proteolytic activity of matrix metalloproteinase 3 on αS. Also, non-enzymatic modification of Lys residues may directly compete with their biological acetylation. αS can be naturally acetylated at its N-terminal amino group, but also on the side chains of K6, K34,K45 and K96. The acetylated forms are seemingly essential for αS to develop a wide range of cellular functions, and aldehydes may hamper αS acetylation and considerably reduce the physiological function of αS.
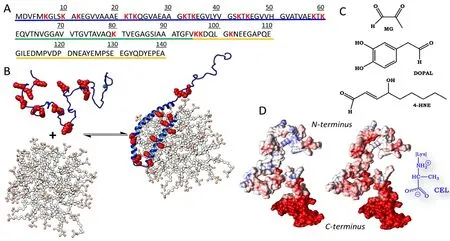
Figure 1 Lys residues act as anchors for α-synuclein (αS) during its binding to lipids, but can also be chemically modified by aldehydes formed in intraneuronal catabolism.
Although Lys residues are seemingly essential for the biological function of αS, their non-enzymatic modification by aldehydes appears to facilitate intraneuronal accumulation of aggregated αS but may also detract from its biological function. These events may increase intraneuronal oxidative stress and alter aldehyde metabolism, thus creating a highly toxic vicious cycle and turning αS into a powerful endotoxin.
This work was supported by the Spanish Government in the framework of Project CTQ2014-55835-R. LM thanks MINECO for award of FPU PhD Grant FPU14/01131.
Ana Belén Uceda, Laura Mariño, Miquel Adrover*
Institut Universitari d’Investigació en Ciències de la Salut (IUNICS),Institut d’Investigació Sanitària Illes Balears (IdISBa), Departament de Química, Universitat de les Illes Balears, Palma de Mallorca,Spain
*Correspondence to:Miquel Adrover, PhD, miquel.adrover@uib.es.
orcid:0000-0002-4211-9013 (Miquel Adrover)
Received:November 15, 2019
Peer review started:November 22, 2019
Accepted:January 22, 2020
Published online:April 3, 2020
doi:10.4103/1673-5374.280309
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Transcriptional regulation of adult neural stem/progenitor cells: tales from the subventricular zone
- Targeting molecular pathways for the treatment of inherited retinal degeneration
- Green tea catechins inhibit microglial activation which prevents the development of neurological disorders
- Reversibility of visual field defects through induction of brain plasticity: vision restoration, recovery and rehabilitation using alternating current stimulation
- Role of activin receptor-like kinase 1 in vascular development and cerebrovascular diseases
- Effects of durotomy versus myelotomy in the repair of spinal cord injury