
伴丘疹性损害的先天性无毛症一例HR基因突变检测
2020-03-16 00:55刘伟英高贵云郑璐瑶
中国麻风皮肤病杂志 2020年1期
刘伟英 高贵云 郑璐瑶 李 明
1湖南航天医院皮肤科,长沙,410205;2上海交通大学医学院附属新华医院皮肤科,上海,200092
先天性无毛症是一组罕见的遗传性皮肤病,具有较强的临床和遗传异质性,其临床表现为头皮或身体其他部位的毛发缺失或完全脱光,伴或不伴有身体其他系统的损害,根据表型可分为综合征型和非综合征型。先天性少毛症的遗传模式常呈常染色体显性遗传、常染色体隐性遗传以及性连锁遗传。伴丘疹性损害的先天性无毛症(atrichia with papular lesions, APL)的遗传模式为常染色体隐性遗传,其致病基因是HR基因[1]。APL患者出生时毛发正常,出生后不久头皮毛发完全脱失且不可逆,大概2岁时,患者开始出现多发的毛囊丘疹,出现在面部、头皮、肢端等,某些患者还可见色素减退斑。本研究中,我们对一例APL患儿进行了HR基因的突变检测,现将结果报道如下。
1 对象与方法
1.1 对象 患儿,女,8岁。足月顺产,出生时毛发与胎脂粘连于头皮,洗澡后头发、眉毛、睫毛大部分脱落,持续加重,头发脱落后不生长,眉毛、睫毛可再生长但不牢固,长至约1 cm后脱落。1岁时患儿头发脱光,近几个月头皮出现散在近肤色丘疹。父母均正常,无兄弟姐妹,家族中其他成员无类似表现。体检:患儿血尿粪常规、肝功能、免疫功能、微量元素测定均无明显异常,智力正常。皮肤科检查:头发、眉毛、睫毛全部脱失(图1a、1b),头皮可见散在近肤色毛囊丘疹(图1c),无掌跖角化,无指(趾)甲异常。
1.2 外周血基因组DNA抽取 征得知情同意后,抽取患儿及其父母共3人的外周静脉血各2 mL。同时采集了100名正常对照的外周血。应用QIA ampDNA Blood Minikit(德国QIAGEN公司)试剂盒提取基因组DNA。
1.3 PCR扩增和测序 采用Primer 5.0软件对HR基因的19个外显子设计特异性引物,进行常规PCR扩增,PCR扩增产物纯化后使用ABI 3730测序仪进行Sanger测序,测序结果使用Chromas软件进行分析,并通过与HR基因cDNA参考序列(NM_005144.4)比较来描述突变。
2 结果
DNA直接测序结果显示HR基因存在复合杂合突变,分别在第10外显子发现移码突变c.2270delC(图2a),导致第757位氨基酸提前终止,变为无义氨基酸。在第15外显子内发现错义突变c.3038T>C(图2b),导致1013位氨基酸由脯氨酸突变为亮氨酸。患儿的两个突变分别遗传于其父亲和母亲。以上2个突变在100名正常对照中均未检测到(图2c、2d),提示它们为致病变异而非单核苷酸多态,查阅国内外文献均属首次报道,在Ensemble、NCBI等数据库均未查到该突变。
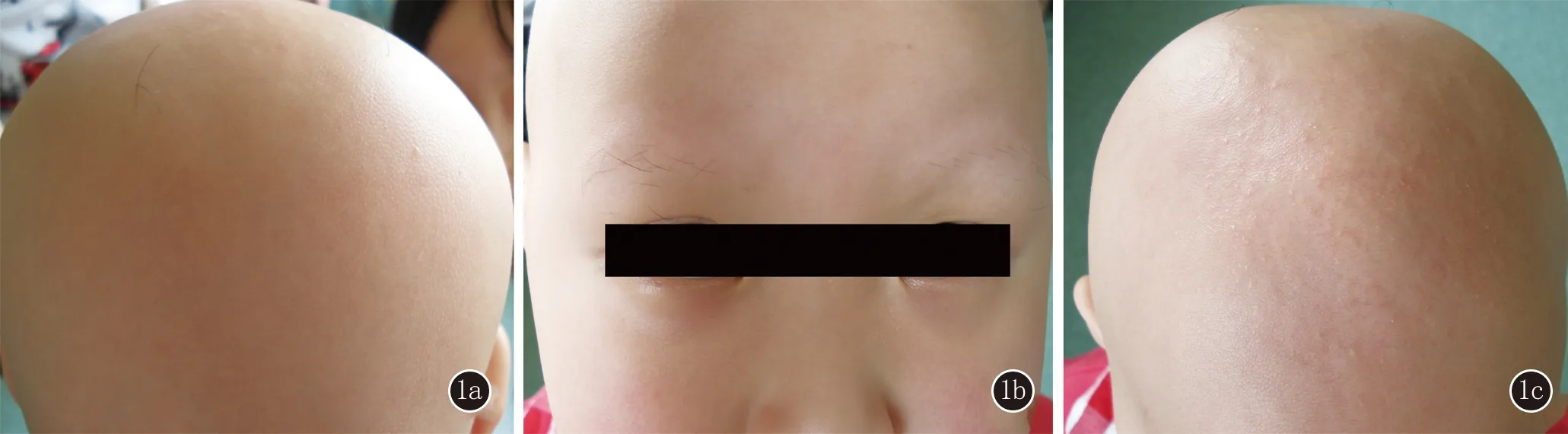
1a、1b:患儿头发和眉毛脱失;1c:头皮可见近肤色的毛囊丘疹
图1临床图片
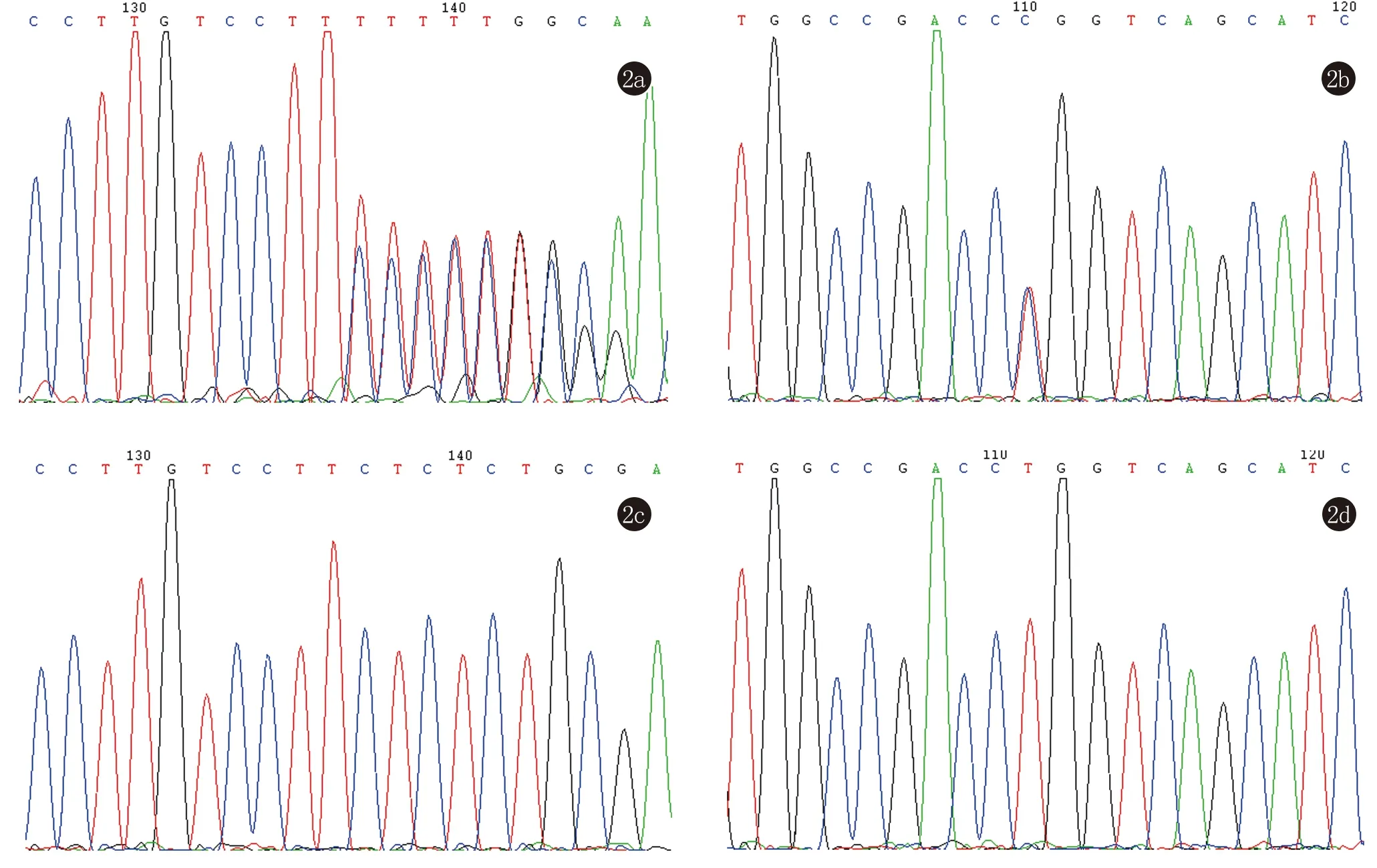
2a:示c.2270delC移码突变;2b:示c.3038 T>C错义突变;2c:正常对照序列;2d:正常对照序列
3 讨论
伴丘疹性损害的先天性无毛症患者临床上可表现为出生时局部或全部的毛发缺失或者出生时毛发正常,在出生后的前几个月脱落,患者还会出现扩散的多发性角化毛囊性丘疹,好发于头皮、面部和四肢。患者无其他外胚层发育的缺陷,如指甲、汗腺或牙齿等,某些患者躯干部可见色素减退斑。APL的遗传模式通常为常染色体隐性遗传。本文报道的患儿为散发病例,出生后不久毛发均脱落,同时伴有丘疹损害,为典型的常染色体隐性遗传APL。
APL通常需要与先天性普秃(alopecia universalis congenita,AUC)进行鉴别诊断,两种疾病均为常染色体隐性遗传,且病因都与无毛基因的突变有关,值得注意的是,AUC患者表现的毛发脱失和APL几乎一致,但前者皮肤是正常的,而APL患者会出现扩散的多角化毛囊性丘疹。仔细询问家族史,根据典型的临床表现不难作出诊断。
1954年,Damste等[2]首次报道了APL,1998年Ahmad等[3]在一个以色列-阿拉伯裔的家系中将该病的致病基因定位于染色体8p21[4],并发现HR基因为本病的致病基因,该基因含有19个外显子,只表达在皮肤和大脑,其表达产物是无毛蛋白,分子量为130kDa,含1189个氨基酸,该蛋白功能的缺陷会导致毛发生长周期的不稳定,其可能在第一个毛发生长周期起作用,由于此蛋白的缺失,毛囊的分化不会被诱导,休止期毛囊不会重新进入生长期,新的毛发也就无法再生长,进而导致毛发的脱落。无毛蛋白功能缺失还可导致毛囊结构的不成熟,从而引起多发性的扩散性毛囊丘疹。
通过对文献及数据库的检索,已经发现了30余种不同的HR基因突变,包括不同的突变类型,如错义突变[5]、无义突变、缺失突变[6]和剪切位点突变[7,8]等。其中国外报道近亲结婚的家系,APL的HR基因的突变均为纯合突变。目前已发现的该基因的突变位点通常分散在整个HR基因上,还未发现热点突变区域。本文报道的患儿家系否认近亲结婚史,患儿携带复合杂合突变,分别为移码突变c.2270delC和错义突变c.3038T>C,在国内外都属首次报道。c.2270delC突变导致基因编码框移码,即p.S757Ffs*144,致使无毛蛋白截短;c.3038T>C引起患者无毛蛋白的第1013位的脯氨酸由亮氨酸替代,即p.P1013L,我们通过Polyphen-2软件(http://genetics.bwh.harvard.edu/pph2/)对其进行功能预测,结果显示该突变很可能是有损害的。这些突变可能导致无毛蛋白的功能异常,导致毛发周期不稳定以及毛囊结构不成熟,最终表现为毛发的脱失以及近肤色的毛囊丘疹。
我们应用HR基因突变检测在1例APL患儿中发现了2种新的HR基因突变c.2270delC和c.3038T>C,增加了中国汉族人群中HR基因的突变谱,为今后进行产前诊断及遗传咨询奠定了基础。
猜你喜欢
医学前沿(2021年6期)2021-09-10
现代临床医学(2021年4期)2021-07-31
感染、炎症、修复(2021年1期)2021-07-28
西南农业学报(2021年1期)2021-05-25
中国生殖健康(2020年2期)2021-01-18
文萃报·周二版(2019年37期)2019-09-10
中华细胞与干细胞杂志(电子版)(2019年1期)2019-05-18
中国生殖健康(2019年11期)2019-01-07
青少年科技博览(中学版)(2017年5期)2018-02-28
小火炬·阅读作文(2014年5期)2015-04-07
