
Promising key genes associated with tumor microenvironments and prognosis of hepatocellular carcinoma
2020-03-12 05:40LongPanJingFangMingYuChenShuTingZhaiBinZhangZhiYuJiangSarunJuengpanichYiFanWangXiuJunCai
Long Pan, Jing Fang, Ming-Yu Chen, Shu-Ting Zhai, Bin Zhang, Zhi-Yu Jiang, Sarun Juengpanich,Yi-Fan Wang, Xiu-Jun Cai
Abstract BACKGROUND Despite significant advances in multimodality treatments, hepatocellular carcinoma (HCC) remains one of the most common malignant tumors.Identification of novel prognostic biomarkers and molecular targets is urgently needed.AIM To identify potential key genes associated with tumor microenvironments and the prognosis of HCC.METHODS The infiltration levels of immune cells and stromal cells were calculated and quantified based on the ESTIMATE algorithm. Differentially expressed genes(DEGs) between high and low groups according to immune or stromal scores were screened using the gene expression profile of HCC patients in The Cancer Genome Atlas and were further linked to the prognosis of HCC. These genes were validated in four independent HCC cohorts. Survival-related key genes were identified by a LASSO Cox regression model.RESULTS HCC patients with a high immune/stromal score had better survival benefits than patients with a low score. A total of 899 DEGs were identified and found to be involved in immune responses and extracellular matrices, 147 of which were associated with overall survival. Subsequently, 52 of 147 survival-related DEGs were validated in additional cohorts. Finally, ten key genes (STSL2, TMC5, DOK5,RASGRP2, NLRC3, KLRB1, CD5L, CFHR3, ADH1C, and UGT2B15) were selected and used to construct a prognostic gene signature, which presented a good performance in predicting overall survival.CONCLUSION This study extracted a list of genes associated with tumor microenvironments and the prognosis of HCC, thereby providing several valuable directions for the prognostic prediction and molecular targeted therapy of HCC in the future.
Key words: Hepatocellular carcinoma; Tumor microenvironment; Differentially expressed genes; Overall survival
INTRODUCTION
Hepatocellular carcinoma (HCC) is a common malignant tumor and the fifth leading cause of cancer-related death in the United States[1]. Due to frequent recurrences and metastases, existing treatments show unsatisfactory efficacy. The pathogenesis of HCC is extremely intricate, involving various biological processes, such as signal transduction (e.g., Ras signaling pathways and WNT/β-catenin pathway) and cell cycle regulation, which displays the interaction among multiple genes in a complex signal network[2,3]. Recently, numerous studies have demonstrated that tumor microenvironments (TMEs) play an important role in cancer development, including HCC, hence affecting the clinical outcomes[4-6]. TMEs consist of immune cells, stromal cells, normal endothelial cells, and vascular cells. Stromal and immune cells are two major components of nontumor cells in the TMEs and are considered to be valuable for the prognostic evaluation of tumors[7,8].
Gene expression profiling has been widely used to identify the potential biomarkers and subsequently construct prognosis models, and it has enriched our knowledge of the underlying molecular mechanisms of tumorigenesis[9-11]. Yoshihara et al[12]designed the ESTIMATE method using gene expression data to calculate the stromal and immune scores, which can predict infiltration levels of two major nontumor cells in tumor issues. The ESTIMATE algorithm has been applied to breast cancer and prostate cancer, showing its feasibility and effectiveness[13,14].
Here, taking advantage of the HCC cohort in The Cancer Genome Atlas (TCGA)database, we used the ESTIMATE method to obtain a list of genes associated with TMEs that predict a poor prognosis in HCC patients. Importantly, such correlation was validated in four different HCC cohorts available from the integrative molecular database of HCC (HCCDB). Key genes were screened by utilizing the LASSO algorithm, and a multigene-based classifier was constructed to further evaluate the relationship between these key genes and the prognosis of HCC.
MATERIALS AND METHODS
Database
The gene expression profile (level 3 data) for HCC patients was acquired from the TCGA data portal (https://xenabrowser.net/datapages/), in which the RNA expression in HCC was analyzed using Illumina HiSeq 2000 RNA Sequencing(October 13, 2017). Clinical data, such as gender, age, grade, tumor stage, survival,and outcome, were also downloaded from TCGA data portal. The ESTIMATE algorithm was used to calculate immune scores and stromal scores, and the score data were obtained from the official data portal (https://bioinformatics.mdanderson.org/estimate/)[12]. The abundances of six types of immune cells (B cells, CD4+ T cells,CD8+ T cells, neutrophils, macrophages, and dendritic cells) in the TCGA were acquired via the TIMER data portal, a web server for comprehensive analysis of tumor-infiltrating immune cells (https:// cistrome.shinyapps.io/timer/)[15,16]. For validation, four HCC datasets were obtained from the HCCDB database(http://lifeome.net/database/hccdb/ home.html), including three Gene Expression Omnibus datasets (HCCDB6, HCCDB7, and HCCDB17) and one Liver Cancer-RIKEN, Japan Project from International Cancer Genome Consortium (HCCDB18)[17].
Identification of differentially expressed genes
Data analysis was conducted using the “limma” package in R[18]. |log2FC| ≥ 1 and FDR < 0.05 were considered statistically significant to screen for differentially expressed genes (DEGs).
Functional enrichment of protein-protein interaction network and module analysis of DEGs
Gene annotation and functional analysis of DEGs were performed by utilizing the“clusterProfiler” in R and Metascape website (a gene annotation & analysis resource)(http://metascape.org)[19,20]. The STRING database was applied to construct potential protein-protein interactions among the DEGs[21]. PPIs with a confidence score ≥ 0.4 were downloaded and reconstructed via Cytoscape[22]. Small networks with less than ten nodes were removed. The connectivity degree of each node in the network was calculated. Moreover, molecular complex detection (MCODE) was performed to detect hub clusters in the PPI network. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were also conducted for significant modules.
Survival analysis
Kaplan-Meier curves were used to estimate the survival differences for patients between the high and low score/risk groups. Univariate Cox proportional hazards regression analyses were conducted to determine the survival-related DEGs. LASSO regression was used to further screen the most powerful prognostic genes. A λ value of 0.066 with log (λ) = -2.72 was selected by 5-fold cross-validation via 1-SE criteria(Supplementary Figure 1). A prognostic gene signature was constructed according to a linear combination of the relative expression values (Expi) and LASSO coefficients(Li) using the following formula: Risk score = L1× Exp1+ L2× Exp2+…+ Ln× Expn. To classify these HCC patients into the high- or low-risk group, the best cutoff value was calculated when the specificity and sensitivity in the receiver operating characteristic(ROC) curve achieved their maximum for predicting 3-year survival using TCGA data. To further evaluate if the prognostic classifier is an independent indicator in patients with HCC, the effect of each clinicopathologic factor on OS was analyzed by univariate Cox regression. Factors whose P value < 0.05 in the univariate analysis were selected for multivariate analysis. Additionally, to test the performance of the prognostic classifier in predicting HCC clinical outcomes, we calculated the area under the curve (AUC) to measure the predictive ability and compared gene signatures with other clinicopathologic features, including TNM stages. All statistical analyses were carried out with R (version 3.6.0) and GraphPad Prism 7 (GraphPad Software Inc., United States).
RESULTS
High immune scores and stromal scores are significantly associated with better prognosis in HCC patients
Gene expression profiles and clinical data of 373 HCC patients with their initial pathologic diagnosis made between 1995 and 2013 were downloaded from the TCGA database. Twenty-nine patients with an overall survival (OS) less than 30 d were excluded. Among the remaining patients, 109 (31.7%) were female, and 235 (68.3%)were male. Based on the ESTIMATE method, immune scores ranged from -1209.16 to 2934.36, and stromal scores were distributed between -1741.56 and 1195.07. Of note,258 patients with scores higher than the 25thpercentile were defined as a “high score group”, while 86 patients with scores lower than the 25th percentile were defined as a“low score group”. The detailed clinical information of HCC patients is shown in Supplementary Table 1. Kaplan-Meier survival curves showed that the mean recurrencefree survival (RFS) and OS in the high-immune score group were better than those in the low-immune score group (RFS: 1032 d vs 478 d, P = 0.0022; OS: 2116 d vs 848 d, P =0. 0272; Figure 1A and B). Patients with high-stromal scores also had consistently better survival benefit in comparison to patients with low-stromal scores (RFS: 990 d vs 384 d, P = 0.0007; OS: 2116 d vs 1005 d, P = 0.0213; Figure 1C and D). Furthermore,detailed information about the infiltration levels of B cells, CD4+ T cells, CD8+ T cells,neutrophils, macrophages, and dendritic cells in the high/low immune score groups is shown in Figure 1E-J (P < 0.0001).
Identification of prognosis-related DEGs in HCC
To explore the survival differences between the high and low score groups, we compared Illumina HiSeq 2000 RNA-seq data of all 373 HCC patients obtained in the TCGA database. For the high and low groups according to immune scores, 1290 genes were downregulated and 48 were upregulated in the low score group (fold change >2; P < 0.05). Similarly, for comparison based on stromal scores, 1649 genes were downregulated and 75 were upregulated in the low score group (fold change > 2; P <0.05). Furthermore, 889 genes were downregulated in both the low immune score group and low stromal score group, while 10 genes were commonly upregulated in the low immune/stromal score group (Figure 2A and B). To investigate the potential roles of individual DEGs in OS, we performed univariate Cox proportional hazards regression analysis using the TCGA database data. Among the 899 DEGs in the low immune/stromal score group, 147 were associated with a significantly poorer prognosis (P < 0.05, Supplementary Table 2). Therefore, we focused on these DEGs for subsequent analyses.
Process and pathway enrichment analysis of prognosis-related DEGs
To summarize the potential functions of 147 DEGs, functional enrichment analyses were performed. GO enrichment analysis indicated the following top GO terms: T cell activation, lymphocyte differentiation, and T cell selection in biological process(Figure 2C); external side of plasma membrane, extracellular matrix, and T cell receptor complex in cell component (Figure 2D); and extracellular matrix structural constituent, carbohydrate binding, and structural molecule activity conferring elasticity in molecular function (Figure 2E). Pathways identified by KEGG analysis were related to the immune response (Figure 2F). Moreover, we performed a network of enriched terms to capture the relationships between the terms by the metascape data portal and found that terms related to the adaptive immune process were in a central position in the network (Figure 2G).
Protein-protein interaction network and module analysis among prognosis-related DEGs
To better explore the interplay among the DEGs of prognostic significance, we generated PPI networks using the STRING data portal. The network consisted of six modules, which included 97 nodes and 420 interactions (Figure 3A). The top module(MCODE score 15.78; 19 nodes and 142 edges) was selected for further analysis(Figure 3B). Significant GO enrichment analysis showed that the module closely correlated with T cell activation, leukocyte cell-cell adhesion, and regulation of leukocyte cell-cell adhesion (Figure 3C). With respect to the KEGG pathway enrichment analysis, DEGs in the module were mainly located in the T cell receptor signaling pathway, cell adhesion molecules (CAMs), and primary immunodeficiency(Figure 3D).
Validation in the HCCDB database
To determine if the genes identified from TCGA-HCC cohort are also of prognostic significance to other HCC-related cohorts, we downloaded the gene expression profile and clinical data of four HCC cohorts from the HCCDB database. A total of 52 genes were validated to be associated with a poor prognosis. Of these genes, 14 were verified by two HCCDB cohorts, and 38were verified by one HCCDB cohort (Figure 4A and B, Table 1). In addition, 38 genes generated particular interest as they have not been reported to be associated with poor outcomes in HCC patients previously.
Construction of prognostic classifier for HCC patients
We utilized LASSO regression to screen ten key genes from the set of 52 genes that are most relevant to OS (Figure 4C, Table 2). The Kaplan-Meier survival curves of ten key genes are shown in Figure 5 and they presented great prediction performance of OS using the TCGA-HCC cohort. The risk score of patients was calculated according to the expression levels of the ten key genes and LASSO coefficients. A total of 251 patients with prognostic scores less than the cutoff value (-1.102) were categorized into the “low-risk group”, while the remaining 93 patients were categorized into the“high-risk group” (Figure 4D and E). An extremely significant difference in OS was detected between the two groups (P < 0.0001, Figure 4F). Figure 4G shows that STSL2 and TMC5 were highly expressed in the high-risk group, whereas the remaining eight genes (DOK5, RASGRP2, NLRC3, KLRB1, CD5L, CFHR3, ADH1C, and UGT2B15) were highly expressly in the low-risk group. To determine if the gene signature is an independent risk factor in patients with HCC, univariate and multivariate Cox regression analyses were conducted. After the multivariable adjustment, the ten-genebased signature remained a significantly independent factor (P = 0.001, Table 3). Inaddition, the prognostic gene classifier showed a good performance in survival prediction, which was better than TNM staging (AUC for 3-year survival: 0.75 vs 0.684, Figure 4H). More importantly, the ten-gene-based signature combined with clinical features, such as Eastern Cooperative Oncology Group performance status and TNM stage, achieved a greater AUC, which was significantly better than the classifier alone. These results suggested that combining the ten-gene-based classifier with clinical features may further improve the ability to predict OS.
DISCUSSION
In this study, low infiltration levels of immune and stromal cells in HCC microenvironments were associated with poor outcomes, and we confirmed the TMErelated genes responsible for OS in the TCGA-HCC cohort. By investigating the gene expression profiles in 373 cases with low vs high immune/stromal scores, 147 prognosis-related DEGs were identified to be involved in immune response and extracellular matrix. More importantly, 52 genes were validated in additional four HCC cohorts from HCCDB, an integrative HCC database containing Gene Expression Omnibus data and International Cancer Genome Consortium data. Ten key genes were screened by the LASSO algorithm and used to construct a prognostic classifier,which showed good performance for predicting OS, indicating a close correlation between these key genes and the prognosis of HCC.
Based on the survival difference between the high and low score groups, 899 DEGs were obtained from the comparison. Among these DEGs, 147 were identified to be related to poor outcomes, many of which are involved in TMEs (Figure 2). These findings are consistent with previous studies showing that TMEs have a critical influence on HCC[4,23,24].
Second, we also generated a PPI network to investigate the correlation among the 147 genes. Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), which was at the center of the network, has been reported to be a tumor suppressor and play a vital role in the tumorigenesis of HCC[25]. Densely connected regions in the network determined by MCODE module analysis are also involved in immune responses and extracellular matrices, which is consistent with the overall enrichment results of all 147 genes. The nodes with a highly connectivity degree in the module containing CXCR6, CCR7, and CD69 have been demonstrated to promote the progression of HCC and be involved in dysfunctions of immune cells[26-28].
By cross validation with four independent HCC cohorts from the HCCDB database,52 TME-related genes were identified, showing a significant correlation between gene expression and OS. Of the 52 genes confirmed, 12 (e.g., CD5, CCL21, CXCR6, GZMA,and SELP) have been reported to be involved in hepatocarcinogenesis or significant in predicting prognosis, indicating that our bioinformatical analysis using TCGA and HCCDB cohorts has reliable prognostic values. The remaining 38 genes have not been previously studied for their effects on hepatocarcinogenesis, suggesting that they may serve as novel therapeutic targets or potential biomarkers for HCC.
Finally, we used LASSO regression analysis to identify ten key genes and subsequently constructed a gene signature showing great capacity for predicting OS.According to our study, CTSL2 and TMC5 were regarded as tumor promoters, while DOK5, CFHR3, ADH1C, UGT2B15, CD5L, RASGRP2, KLRB1, and NLRC3 wereregarded as tumor suppressors. NLRC3, a member of the nucleotide-binding domain and leucine-rich repeat (NLR) family protein, plays an important role in immunity and inflammation[29,30]. Ma et al[31]reported that high expression levels of NLRC3 correlate with a favorable clinical prognosis in a Chinese HCC population and that downregulation of NLRC3 expression inhibits cell apoptosis and contributes to cell proliferation in vitro. Maehara et al[32]investigated CD5L, which is the apoptosis inhibitor of macrophages, and they found that CD5L prevents hepatocellular carcinoma through complement activation. Two other studies have also shown that CD5L favors overall HCC survival, which agreed with our study[33,34]. Therefore, the prognostic value of the additional eight key genes reported in this study may provide a valuable direction for future research.

Table 2 Top 10 genes predicting overall survival screened by lasso regression in hepatocellular carcinoma cohort from The Cancer Genome Atlas
The interactions of HCC and the TME significantly affect tumor evolution, hence influencing tumor escape, tumor recurrence, drug resistance, and patient clinical outcomes[4,35-37]. Remarkable progress has been made in the research of the interplay between HCC and TME, and most of these experiments have been performed in cell lines, animal models, or small cohorts based on patient samples. However, the complexity of HCC and TME requires additional comprehensive analysis.Fortunately, the development of tumor databases, including TCGA and HCCDB, has provided a solid foundation for high-throughput screening of large-scale HCC cohorts. Our results provide novel data revealing the complex interplay of tumor and TME in HCC.
There are several limitations in our study. First, due to the current study being implemented based on bioinformatics analysis, biological experiments are urgently needed for validation. Second, as the data used were from public databases, their quality was not effectively assessed. Third, the clinical characteristic of patients were not taken into account in survival analysis since identification of the prognosis-related genes from multiple datasets was the primary focus.
In conclusion, this study extracted a series of genes associated with TME and prognosis from the TCGA database using the ESTIMATE method. These genes were validated in four independent HCC cohorts. After screening by LASSO regression, ten key genes were used to construct a gene signature, showing good performance in predicting OS. These findings contribute to outlining the prognosis and molecular targeted therapy of HCC. Further investigations of these key genes may bring a novel insight into the potential association of TME with HCC prognosis in a more comprehensive pattern.
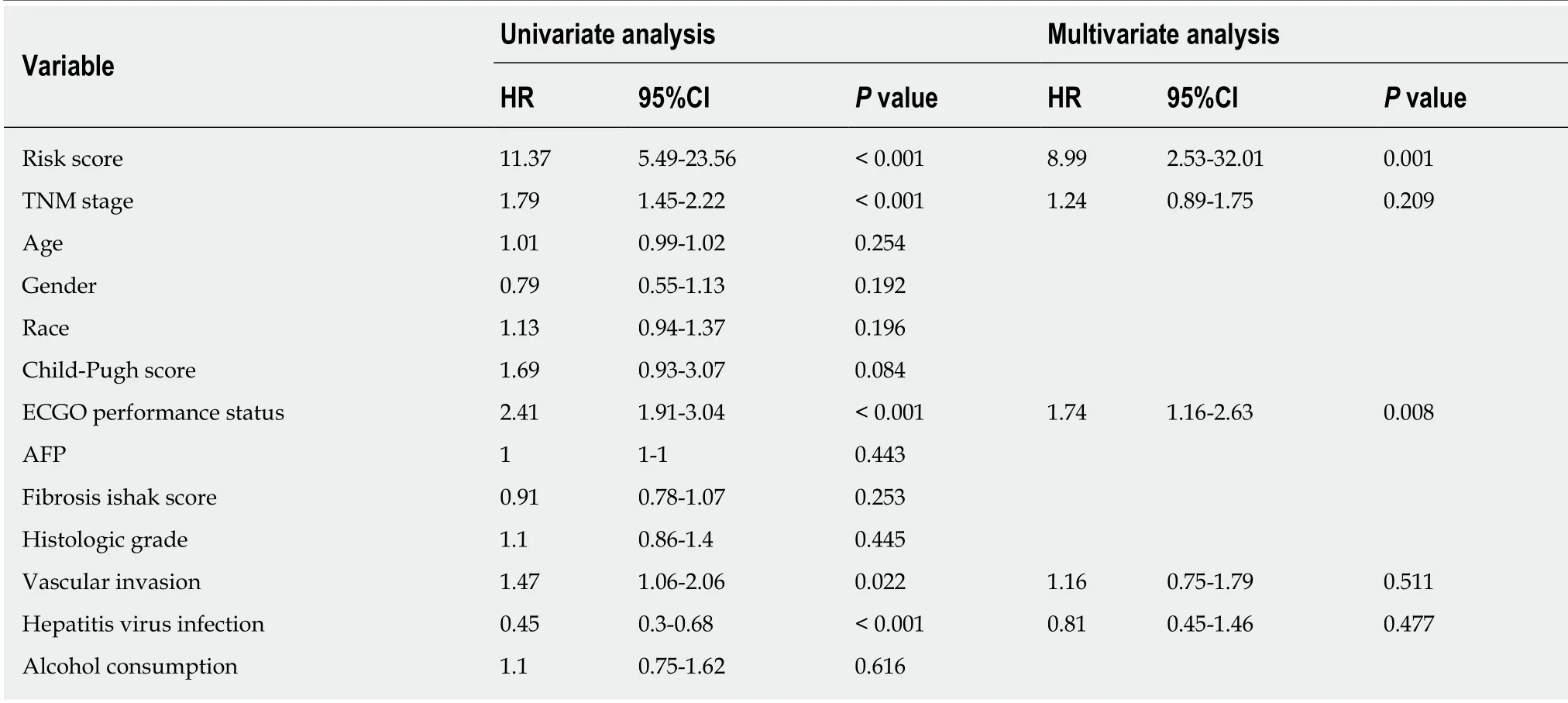
Table 3 Univariate and multivariate Cox regression analyese of prognostic factors and overall survival of patients with hepatocellular carcinoma from The Cancer Genome Atlas database

Figure 2 Pathway and process enrichment analysis of prognosis-related differentially expressed genes in hapetocellular carcinoma. A and B: Venn diagrams showing the number of simutaneously downregulated (A) or upregulated (B) differentially expressed genes (DEGs) in immune or stromal score groups; C-F:Gene ontology terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis across 147 DEGs with regard to (C) biological process, (D) cellular component, (E) molecular function, and (F) KEGG pathway. G: Network of enriched terms. Nodes that share the same terms are typically close to each other. KEGG:Kyoto Encyclopedia of Genes and Genomes.

Figure 3 Protein-protein interaction network and module analysis of prognosis-related differentially expressed genes. A: Protein-protein interaction (PPI)network of all 147 differentially expressed genes; B: PPI network of the module. The color of nodes in the PPI network reflects the log (FC) value of the Z score of gene expression, and the size of nodes means the number of interacting proteins with the designated protein; C and D: Gene ontology terms and KEGG pathway analysis for the genes in the module. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
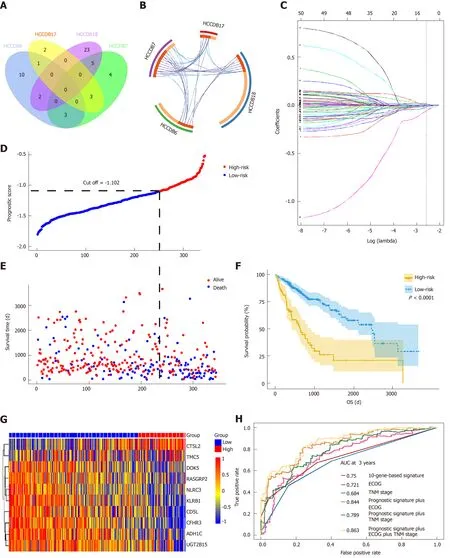
Figure 4 Prognosis-related differentially expressed genes validated in four additional cohorts and construction of the prognostic gene signature. A: Venn diagram showing the results of genes validated in four cohorts; B: Circos plot showing overlapping genes between the four cohorts. Purple curves link identical genes while blue curves link genes that belong to the same enriched ontology term; C: LASSO coefficient profiles of 52 genes validated in four cohorts; D: Distribution of risk scores; E: Patients' survival time and status. The black dotted line indicates the optimum cutoff dividing patients into low- and high-risk groups; (F) Kaplan-Meier curves for low- and high-risk groups; G: Heat map of ten key genes in the genes signature; H: Time-dependent receiver operating characteristic curves for comparing prediction accuracy among the ten-gene-based signature and clinicopathological features.


Figure 5 Correlation of expression of the ten genes with overall survival in The Cancer Genome Atlas. Kaplan-Meier survival curves were generated by using the website “Kaplan Meier plotter” (http://kmplot.com/analysis). The the best performing threshold was selected as a cutoff.
ARTICLE HIGHLIGHTS
Research background
Tumor microenvironments (TMEs) play an important role in cancer development, including hepatocellular carcinoma (HCC), hence affecting clinical outcomes. Stromal and immune cells are two major components of nontumor cells in TMEs and are considered to be valuable for the prognostic evaluation of tumors. Nonetheless, the infiltration level of stromal/immune cells and specific roles of TME-related genes in HCC have not yet been clarified.
Research motivation
More biomarkers are required for the diagnosis and treatment of HCC.
Research objectives
The present study investigated potential key genes associated with tumor microenvironments and the prognosis of HCC.
Research methods
The ESTIMATE method was used to predict the infiltration levels of stromal and immune cells in HCC. Differentially expressed genes (DEGs) between patients with high and low infiltration levels were screened using the TCGA database and linked to overall survival of HCC. DEGs were verified by four other independent HCC cohorts and further selected by LASSO Cox regression analysis.
Research results
HCC patients with high immune/stromal infiltration levels had better survival benefits than patients with low infiltration levels. A total of 147 DEGs were identified to be associated with overall survival. Moreover, 52 survival-related DEGs were validated, and ten key genes were selected by LASSO algorithm, which were further used to construct a prognostic classifier,showing a good performance in predicting overall survival.
Research conclusions
Our study screened a series of TME-related genes and provided a novel insight into the potential association of TME with HCC prognosis and molecular targeted therapy of HCC in the future.
Research perspectives
HCC and TMEs are an integral whole. More in-depth research should be conducted to reveal the important role of components of TMEs in HCC to develop novel anti-cancer treatments via targeting TME-related cells and the extracellular matrix.
ACKNOWLEDGEMENTS
We thank Yi-Dong Pan (Guy's, King's & St Thomas's (GKT) School of Medical Education, King's College London, London, United Kingdom) and AJE(www.aje.com) for their linguistic assistance during the preparation of this manuscript.
 World Journal of Gastroenterology2020年8期
World Journal of Gastroenterology2020年8期
- World Journal of Gastroenterology的其它文章
- Therapies to modulate gut microbiota: Past, present and future
- Downregulation of orosomucoid 2 acts as a prognostic factor associated with cancer-promoting pathways in liver cancer
- Neoadjuvant chemotherapy vs upfront surgery for gastric signet ring cell carcinoma: A retrospective,propensity score-matched study
- Haemoglobin, albumin, lymphocyte and platelet predicts postoperative survival in pancreatic cancer
- Prognostic value of preoperative weight loss-adjusted body mass index on survival after esophagectomy for esophageal squamous cell carcinoma
- Diverting colostomy is an effective and reversible option for severe hemorrhagic radiation proctopathy