
同位素内标-气相色谱-质谱法同时测定婴幼儿配方粉中3-氯丙醇酯和缩水甘油酯的含量
2020-03-11 08:40高红波隋海霞钟其顶吕晓玲
食品科学 2020年4期
王 亚,高红波,隋海霞,仇 凯,*,钟其顶,吕晓玲*
(1.天津科技大学食品科学与工程学院,天津 300457;2.中国食品发酵工业研究院,北京 100015;3.全国食品发酵标准化中心,北京 100015;4.国家食品安全风险评估中心,北京 100022)
脂肪酸氯丙醇酯(简称氯丙醇酯)污染是近年来国际上的食品安全研究热点。氯丙醇酯按照氯丙醇种类的不同主要分为4 种[1],其中污染水平最高的是3-氯丙醇酯(3-monochloropropane-1,2-diol esters,3-MCPDE)[2]。3-MCPDE的毒性至今仍不清楚,可能引起健康风险的主要原因是3-MCPDE在加热、酸性等条件下会水解成游离的3-氯-1,2-丙二醇(3-monochloropropane-1,2-diol,3-MCPD)[3],联合国粮农组织/世界卫生组织食品添加剂委员会指出3-MCPD是一种具有肾脏、生殖和神经毒性的致癌物[4-5],同时3-MCPD也被国际癌症组织划分为2B类致癌物[6]。缩水甘油酯(glycidol esters,GEs)是缩水甘油和游离脂肪酸的酯化产物,同时也是一种新型的油脂危害物。2010年,德国联邦风险评估研究所通过毒理学实验证实了GEs本身并不具有致癌性,但其进入人体代谢后所产生的缩水甘油具有遗传毒性[5]。GEs被国际癌症组织定义为人类2A级致癌物[7],同时它也被德国油脂科学协会定义为2类致癌物[5]。2018年,欧盟委员会条例规定截止至2019年6月30日,婴幼儿配方奶粉、较大婴儿配方奶粉和婴幼儿特殊医用食品(粉状)中GEs的最高限量为75 μg/kg,从2019年7月1日起,其GEs的最高限量为50 μg/kg。
目前关于食品中氯丙醇酯的检测方法主要分为直接检测法和间接检测法,如直接检测法中的高效液相色谱-飞行时间质谱[8-9],间接检测法中的气相色谱或气相色谱-质谱(gas chromatography-mass spectrometry,GC-MS)联用[10-12]。现行的GB 5009.191ü2016《食品中氯丙醇及其脂肪酸酯含量的测定》第三法食品中氯丙醇脂肪酸酯含量的测定GC-MS法,也是一种间接检测法,该方法多用于食用植物油中氯丙醇酯的测定[13-14]。GEs具有一定的热不稳定性,因此对其检测也有一定的难度。目前关于食品中GEs的检测分为直接检测法和间接检测法,如直接检测法中的双固相萃取-三重四极杆液相色谱-质谱直接测定[15],间接检测法中的GC或GC-MS[16-21]。对比2 种物质的直接检测法和间接检测法,直接检测法的测定时间短,但所需的标准品多,价格昂贵,而间接检测法所需的标准品少,测定的灵敏度高,多用于常规测定。
婴幼儿配方粉是非母乳喂养婴儿的主要营养素摄入来源,3-MCPDE与GEs是婴幼儿食品中潜在的危害物质,欧盟已建立相关限量标准,我国尚未建立相关限量标准,因此建立婴幼儿配方粉中3-MCPDE和GEs的检测方法对于开展风险评估及标准制定具有重要意义。目前有关食品中3-MCPDE和GEs同时测定的间接检测方法较少[22],被检测的样品也以食用油为主,关于婴幼儿配方粉中3-MCPDE和GEs同时测定的检测方法以及这2 种物质的污染源解析和健康风险研究几乎处于空白阶段,本研究以婴幼儿配方粉为研究对象,建立一种同时测定3-MCPDE和GEs含量的间接检测法,样品经提取、转化、碱性水解、苯硼酸衍生化4 步前处理,最终经GC-MS联用仪测定,并采用该方法对我国市售部分婴幼儿配方奶粉进行检测。本研究拟为婴幼儿配方粉中3-MCPDE和GEs检测方法的完善和相关标准的制定提供参考,为婴幼儿配方粉中3-MCPDE和GEs的污染源解析和健康风险研究提供新的解决办法。
1 材料与方法
1.1 材料与试剂
市购婴幼儿配方粉样品:20 个品牌共30 种婴幼儿配方粉样品,其中包括I段婴幼儿配方粉样品24 种,II段婴幼儿配方粉样品4 种,III段婴幼儿配方粉样品2 种。
顺式-3-氯-1,2-丙二醇棕榈酸二酯(简称3-MCPD棕榈酸二酯,纯度98.0%)、棕榈酸缩水甘油酯(纯度98.0%)、顺式-3-氯-1,2-丙二醇棕榈酸单酯-D5(简称D5-3-MCPD棕榈酸单酯,纯度99.0%)、3-MCPD(纯度98.0%)、3-甲氧基丙烷-1,2-二醇(3-methoxypropane-1,2-diol,3-MPD,纯度98.0%)、3-氯-1,2-丙二醇-D5(3-MCPD-D5,纯度97.0%)、甲基叔丁基醚(methyl tert-butyl ether,MTBE,纯度99.0%)、苯基硼酸(phenylboronic acid,PBA,纯度98.0%)、甲醇钠(纯度99.0%) 北京百灵威科技有限公司;丙酮(纯度99.8%)、正己烷(纯度95.0%)、甲醇(纯度99.9%)、乙酸乙酯(纯度99.9%) 韩国Duksan Pure Chemicals公司;无水硫酸钠(纯度99.0%)、硫酸(纯度98%)、氯化钠(纯度99.5%)、冰醋酸(纯度99.5%)(均为分析纯) 北京化工厂;实验用水均为去离子水。
1.2 仪器与设备
2010 Plus-QP 2020 GC-MS仪 日本岛津公司;XS205电子分析天平 梅特勒-托利多仪器(上海)有限公司;MX-S涡旋振荡器 大龙兴创实验仪器(北京)有限公司;HC-3518高速离心机 安徽中科中佳科学仪器有限公司;DF-101S集热式恒温加热磁力搅拌器 巩义市予华仪器有限责任公司。
1.3 方法
1.3.1 标准溶液的配制
D5-3-MCPD棕榈酸单酯标准储备液(1 000 mg/L,以对应的氘代氯丙醇计):准确称取D5-3-MCPD棕榈酸单酯标准品30.60 mg(精确到0.01 mg)用乙酸乙酯溶解,转移至10 mL容量瓶中,用乙酸乙酯定容至刻度,混匀。
3-MCPD棕榈酸二酯标准储备液(1 000 mg/L,以对应的氯丙醇计):准确称取3-MCPD棕榈酸二酯标准品53.10 mg(精确到0.01 mg)用乙酸乙酯溶解,转移至10 mL容量瓶中,用乙酸乙酯定容至刻度,混匀。
棕榈酸缩水甘油酯标准储备液(1 000 mg/L):准确称取棕榈酸缩水甘油酯标准品10.00 mg(精确至0.01 mg),用乙酸乙酯溶解,转移至10 mL容量瓶中,用乙酸乙酯定容至刻度,混匀。
3-MCPD-D5标准储备液(1 000 mg/L):准确称取3-MCPD-D5标准品10.00 mg(精确至0.01 mg),用乙酸乙酯溶解,转移至10 mL容量瓶中,用乙酸乙酯定容至刻度,混匀。
3-MCPD标准储备液(1 000 mg/L):准确称取3-MCPD标准品10.00 mg(精确至0.01 mg),用乙酸乙酯溶解,转移至10 mL容量瓶中,用乙酸乙酯定容至刻度,混匀。
3-MPD标准储备液(1 000 mg/L):准确称取3-MPD标准品10.00 mg(精确至0.01 mg),用无水乙醇溶解,转移至10 mL容量瓶中,用无水乙醇定容至刻度,混匀。
标准工作液(1 0 m g/L):移取标准储备液(1 000 mg/L)0.1 mL于10 mL容量瓶中,用MTBE定容至刻度线,混匀,得到所需的各种标准工作液。
系列标准混合工作液:分别吸取3-MCPD和3-MPD混合标准工作液0.01、0.02、0.04、0.08、0.16 mL于10 mL离心管中,加入0.15 mL的3-MCPD-D5标准工作液,加20%的NaCl溶液至2 mL,混匀,临用现配。
1.3.2 样品前处理
1.3.2.1 样品提取
准确称取奶粉样品1.00 g(精确到0.01 g)于50 mL离心管中,加入6 mL 65 ℃温水匀浆后,再加入0.15 mL的D5-3-MCPD棕榈酸单酯标准工作液。混匀后加入10 mL MTBE与乙酸乙酯混合溶液(8∶2,V/V)提取,涡旋振荡5 min,8 000 r/min离心5 min取上清液。按上述步骤提取3 次,将3 次上清液合并,于45 ℃条件下氮吹浓缩至溶液体积约为2 mL。
1.3.2.2 GEs的转化
向上述2 mL提取液中加入1 mL 1%硫酸-甲醇溶液,将混合溶液充分混匀后在50 ℃水浴中加热30 min。
1.3.2.3 酯类水解
将上述混合溶液冷却至室温后加入200 µL甲醇钠-甲醇溶液(0.25 g/mL)裂解3-MCPDE和3-甲氧基丙烷-1,2-二醇酯。密闭条件下,将混合溶液充分振荡。反应60 s后,加入200 μL冰醋酸和2 mL 20%的NaCl溶液终止反应。充分振荡完成分析物的提取,待两相分离后,弃去上层有机相。
1.3.2.4 衍生化反应
将200 µL衍生化试剂PBA加入水层中,充分振荡。在室温下反应15 min后,加入2 mL正己烷涡旋振荡1 min。取正己烷层,加入0.3 g无水硫酸钠,将脱水后的正己烷溶液转移至进样小瓶中,供GC-MS测定分析。同时做系列标准混合工作液的衍生化反应。
1.3.3 GC-MS测定条件
G C条件:S H-R x i-5 m s毛细管色谱柱(30 mh0.25 mm,0.25 µm);程序升温:初始温度50 ℃,保持1 min,以10 ℃/min升温至210 ℃,再以30 ℃/min升温至300 ℃,保持5 min;总采集时间25 min;进样口温度250 ℃;载气为超纯级氦气,流速0.7 mL/min;不分流进样;进样量2 µL。
MS条件:电子电离源;电离能量70 eV;离子源温度250 ℃;传输线温度280 ℃;采用选择离子监测方式。
1.3.4 有机溶剂提取率的计算
移取0.1 mL 3-MCPDE标准中间液,加入0.9 mL有机溶剂,充分振荡后,加入无水硫酸钠吸水干燥,将溶液转移至进样小瓶中,供GC测定分析,记其目标化合物的响应值为A1;移取0.1 mL 3-MCPDE标准中间液于0.5 mL温水混匀后的婴幼儿配方粉样品溶液中,加入0.9 mL有机溶剂,充分振荡后分离有机层,再经上述相同的操作后,记其目标化合物的响应值为A2。提取率计算如下式所示:
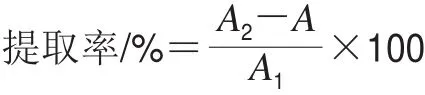
式中:A为婴幼儿配方粉样品中3-MCPDE的本底值。
1.4 数据处理
利用岛津GC Solution软件对数据进行采集,使用Origin 8.0软件绘图。
2 结果与分析
2.1 样品前处理的优化
2.1.1 3 种衍生物的定性分析
经苯硼酸衍生后3 种目标化合物的衍生物可以通过SH-Rxi-5 ms毛细管色谱柱实现分离,且目标化合物的保留时间、峰形均良好,可以达到很好的分离效果。在最优的色谱条件下,3 种目标化合物的总离子流色谱图如图1所示,保留时间和监测离子对如表1所示。3-MPD、3-MCPD衍生物的m/z147和3-MCPD-D5衍生物的m/z150灵敏度最高,因此选择m/z147作为3-MPD、3-MCPD衍生物的定量离子,选择m/z150作为3-MCPD-D5衍生物的定量离子,其他离子作为各目标化合物的定性离子。

图1 3-MCPD和3-MPD衍生物及内标衍生物的总离子流色谱图Fig. 1 Total ion current chromatograms of derivatives of 3-MCPD,3-MPD and internal standard
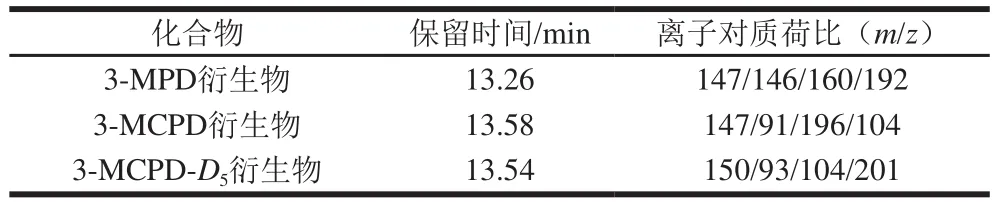
表1 目标化合物的色谱及质谱参数Table 1 Mass spectrometric parameters for target compounds
2.1.2 提取溶剂的选择
目标化合物的提取是婴幼儿配方粉中3-MCPDE和GEs检测的前提。采用GB 5009.191ü2016第三法食品中氯丙醇脂肪酸酯含量的测定GC-MS法中的提取方法,经过本实验室的验证,正己烷的提取效率较低。本研究对比正己烷[1,23]、MTBE[24]、乙酸乙酯[1]以及MTBE-乙酸乙酯的混合溶液(8∶2,V/V)[25]的提取效果,结果如表2所示,选用MTBE与乙酸乙酯的混合溶液作为提取剂最佳。因此采用MTBE和乙酸乙酯的混合溶液作为本实验的提取试剂。

表2 不同有机溶剂的提取率Table 2 Extraction efficiencies with different organic solvents
2.1.3 转化条件的优化

图2 实验过程中物质转化流程图Fig. 2 Flow chart of material conversion during the experiment
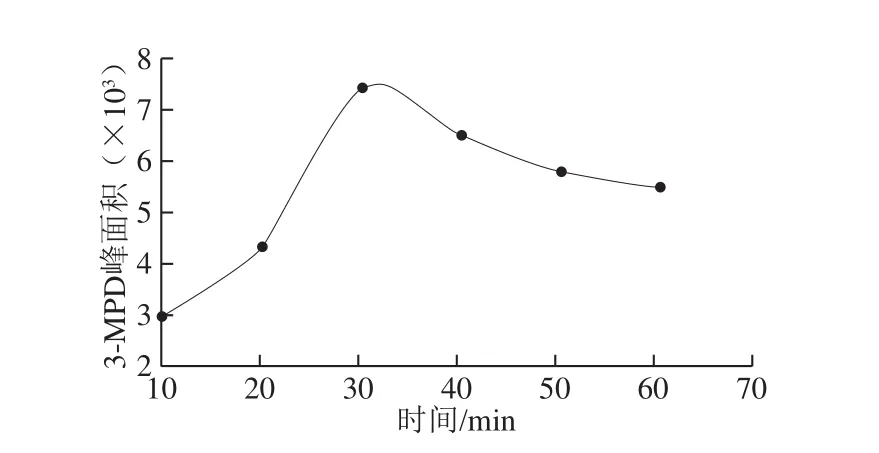
图3 转化时间对目标化合物响应值的影响Fig. 3 Effect of conversion time on response value of target compounds
在酯交换过程中甲醇钠通过分子内的亲核取代使R-卤代醇,即3-MCPD转化成大量的缩水甘油,从而导致3-MCPD的含量减少而缩水甘油的含量增加[26-28],为避免副反应的产生,本实验采用酸性醇解实现3-MCPDE和GEs的明显区分(图2),并通过转化产物实现准确定量。缩水甘油与硫酸-甲醇溶液的开环反应有高度的区域选择性,主要形成3-MPD,且反应具有良好的再现性[24]。将转化产物3-MPD作为参比化合物,发现酸性醇解实现了3-MCPDE和GEs的明显区分。文献[24]提出50 ℃为转化反应的最佳温度,更高的温度会导致转化率降低且有大量副反应产生,为提高转化率,本研究以10、20、30、40、50、60 min 6 个时间水平为变化因素,以目标化合物的响应值为评价指标进行单因素试验,如图3所示。10~30 min时,目标化合物峰面积增大,缩水甘油开环逐渐转化为3-MPD;30 min后,目标化合物峰面积减小,转化率降低,故采用30 min作为转化反应的时间。
2.1.4 水解时间的确定
由于3-MCPD在碱性介质中容易降解,故水解时间对该方法的灵敏度具有很大影响[29]。为减少副反应的产生,在保证酯类完全水解的基础上,应使水解反应的时间尽可能短[24]。以水解时间40、50、60、70、80 s和90 s进行水解反应优化实验条件,结果如图4所示。40~60 s,目标化合物峰面积增大,酯类逐渐水解,且在水解过程中3-MPD单酯的水解速率与3-MCPD双酯的水解速率相比较快。60 s之后,目标化合物峰面积减小,3-MCPD在碱性条件下降解,且二者的降解速率趋于一致。酯类完全裂解且副反应最少的反应时间为60 s,故本实验采用60 s作为水解时间。
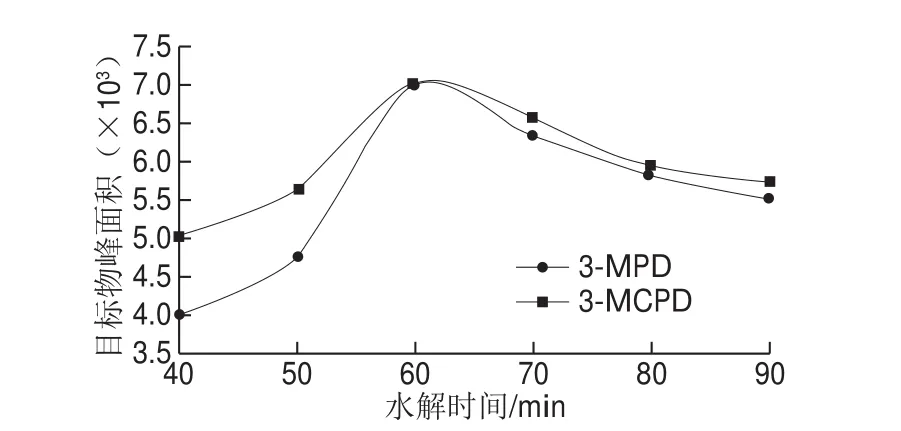
图4 水解时间对目标化合物响应值的影响Fig. 4 Effect of hydrolysis time on response value of target compounds
2.2 线性方程和检出限结果
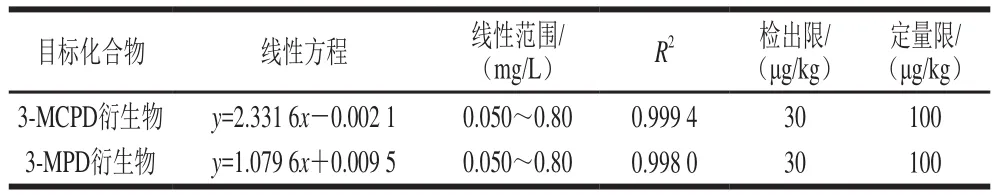
表3 目标化合物的线性方程、线性范围、相关系数(R2)、检出限和定量限Table 3 Standard curves, linear ranges, correlation coefficients (R2),LODs and LOQs of target compounds
分别以3-MCPD衍生物色谱峰面积与内标衍生物的色谱峰面积之比为纵坐标,3-MCPD质量浓度为横坐标,以3-MPD衍生物的色谱峰面积与内标衍生物的色谱峰面积之比为纵坐标,3-MPD质量浓度为横坐标,绘制标准曲线,得到目标化合物在0.050~0.80 mg/L质量浓度范围内的线性方程,以3 倍信噪比计算方法的检出限,以10 倍信噪比计算方法的定量限,结果如表3所示。经验证目标化合物在0.050~0.80 mg/L质量浓度范围内线性关系良好,R2均大于0.998,检出限和定量限分别为30 μg/kg和100 μg/kg。
2.3 回收率和精密度测定结果
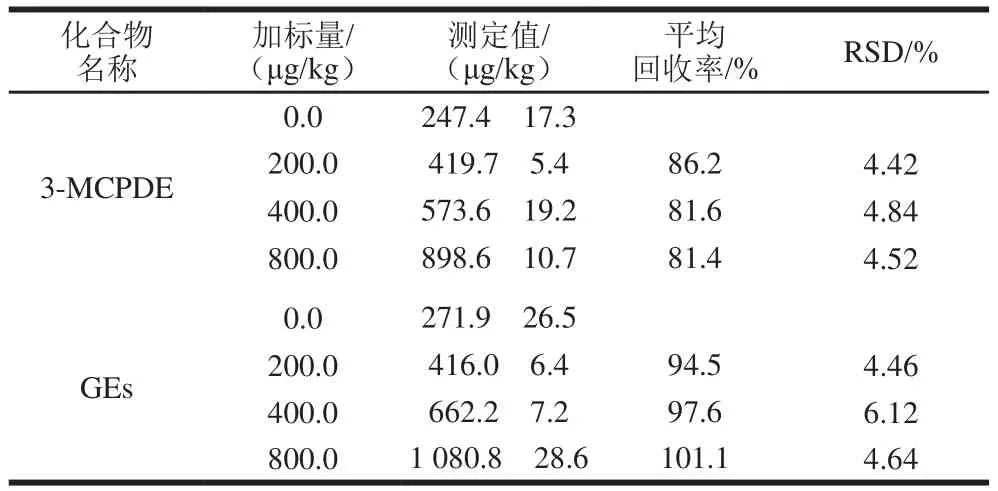
表4 样品的加标回收率及精密度(n=6)Table 4 Recoveries and precisions for spiked samples (n= 6)
在最优实验条件下,选取样品分别加入200.0、400.0 μg/kg和800.0 μg/kg 3 个添加量的3-MCPDE和GEs混合标准工作液,每个添加水平平行测定6 次,计算平均回收率和相对标准偏差(relative standard deviation,RSD)。由表4可知,当样品中目标化合物的加标水平在200.0~800.0 μg/kg时,平均回收率在81.4%~101.1%,RSD为4.42%~6.12%,表明方法的精密度良好,能够满足婴幼儿配方粉中3-MCPDE和GEs的测定要求。
2.4 市售样品分析评价
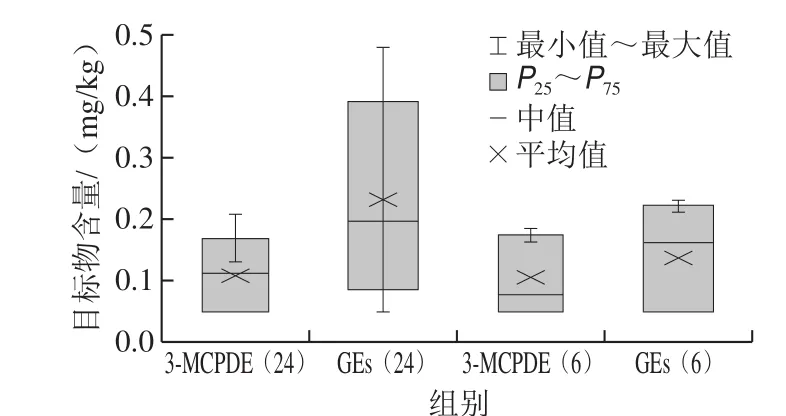
图5 2 种目标化合物含量分析箱线图Fig. 5 Box plots of the contents of two target compounds
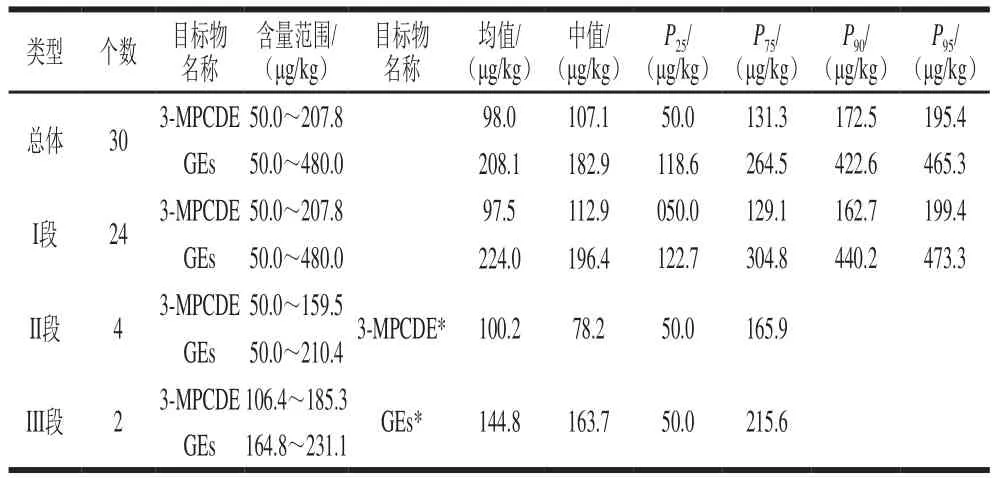
表5 不同阶段婴幼儿配方粉样品中3-MCPDE和GEs含量分析Table 5 Analysis of 3-MCPDE and GEs contents in infant formula powder samples intended for different age groups
本研究对30 份我国市售婴幼儿配方粉样品中3-MCPDE和GEs的含量进行测定,每份样品做2 个平行,取平均值,结果见表5。30 份样品中,3-MCPDE和GEs的检出率为100%,而且从图5可以看出,II段、III段婴幼儿配方粉中3-MCPDE和GEs的含量低于I段婴幼儿配方粉中目标化合物含量的均值与中值。一般认为3-MCPDE和GEs来源于脂肪,而II段、III段婴幼儿配方粉中脂肪含量相对于I段奶粉中脂肪的含量会有所减少,由此可见婴幼儿配方粉中这2 种目标化合物的含量可能与脂肪含量的多少有一定的相关性,同时还可能与配方粉中所添加的食用植物油的种类[30]等有关。
3 结 论
本研究利用同位素内标和GC-MS法建立婴幼儿配方粉中3-MCPDE和GEs的同步检测方法,实验对提取溶剂的选择、酸性醇解的转化条件和酯类水解的时间进行了优化。方法学评价表明,3-MCPDE和GEs在0.050~0.80 mg/L范围内的线性关系良好,方法的检出限均达到30 μg/kg。样品基质加标回收率测定结果表明,该方法回收率高,精密度良好,能满足不同阶段婴幼儿配方粉中3-MCPDE和GEs的定量检测要求。本实验通过对大量样品的测定可知:30 份婴幼儿配方粉样品中3-MCPDE含量为50.0~207.8 μg/kg,平均值为98.0 μg/kg,GEs含量为50.0~480.0 μg/kg,平均值为208.1 μg/kg。本研究为完善其他食品中3-MCPDE和GEs的同步检测方法提供参考,为婴幼儿配方粉中3-MCPDE和GEs检测方法的完善和相关标准的制定提供指导,为婴幼儿配方粉中3-MCPDE和GEs的污染源解析和健康风险研究提供了新的解决办法。
猜你喜欢
分子催化(2022年1期)2022-11-02
食品工业科技(2021年23期)2021-12-16
建材发展导向(2021年19期)2021-12-06
昆明医科大学学报(2021年8期)2021-08-13
统计与决策(2019年7期)2019-05-05
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中国油脂(2017年7期)2017-09-16
食品与生活(2017年5期)2017-05-27
哈尔滨理工大学学报(2016年3期)2016-11-05
