
韩日澳三国兽用生物制品批签发管理简介
2020-03-05 06:43张存帅
中国兽医杂志 2020年10期
娜 琳,李 宁,李 琰,张存帅
(中国兽医药品监察所,北京 海淀 100081)
兽用生物制品是指用于预防、治疗、诊断畜禽等动物特定传染病或其他有关的疾病的菌苗、疫苗、虫苗、类毒素、诊断制剂和抗血清等制品。兽用生物制品的质量直接关系到疫病预防效果,从而间接影响到公共卫生和环境安全等。全球许多国家都对兽用生物制品施行批签发管理,但依据不同国情,管理方式也各不相同。笔者在此对韩国、日本和澳大利亚的兽用生物制品管理进行概述,同时分析各国的特点及与我国管理方面的区别,为我国进一步做好兽用生物制品的批签发提供参考。
1 韩国兽用生物制品批签发的管理情况
1.1 兽用生物制品批签发管理现状 韩国的兽用生物制品批签发系指由韩国农林水产食品部下属的农林畜产检疫本部对兽用生物制品做出是否准予上市的行政审批。韩国对兽用生物制品实施批签发管理开始于1966年1月11日,批签发抽样、检验等具体工作由农林畜产检疫本部下属的动物药品管理科和动物药品评价科负责。韩国的兽用生物制品批签发法律依据是《韩国兽药管理法规及指导原则》。2018年韩国的兽用生物制品批签发总量为1 300批,其中进行批签发检验的约为300批[1]。
1.2 需要批签发的产品种类 所有的兽用疫苗在上市前均需进行批签发,诊断试剂则不需进行批签发。
1.3 兽用生物制品批签发流程 韩国的兽用生物制品批签发设立了免检制度,如果某个产品连续10批批签发检验均合格(没有年限要求),则该产品可以被批准免检。如某一批产品上市后出现相关质量问题,则会取消该产品的免检资格,直到再次有10个批次产品的批签发检验均合格,才能重新被批准免检。企业提出批签发申请后,如果是免检的产品,则仅进行资料审查;如果是需要检验的产品,则企业需缴纳相应的检验检测费用,由动物药品管理科人员到企业进行抽样,并将封存的样品交给动物药品评价科进行检验。对于不予签发的产品,由动物药品管理科监督企业对该批产品进行销毁,流程见图1。

图1 韩国兽用生物制品批签发流程
1.4 兽用生物制品批签发抽样检验 韩国的兽用生物制品批签发抽样由动物药品管理科负责,检验由动物药品评价科负责。动物药品管理科人员在进行抽样时,会将留样封存在企业,动物药品管理科和动物药品评价科均不会保存留样。批签发检验的时限为3个月。检验项目根据不同的产品有所不同,主要的关注点在于产品的安全性和有效性,如理化特性、性状、半数致死量、抗原含量、抗体效价等。如果检验不合格,企业可在批签发结果公布当天起14天内向动物药品管理科申请1次复检。
1.5 批签发核查和检查 韩国不对产品进行批签发现场核查,而是通过兽药生产质量管理规范(GMP)检查对产品质量进行控制。
2 日本兽用生物制品批签发的管理情况
2.1 兽用生物制品批签发管理现状 日本的兽用生物制品批签发是指由日本农林水产省(MAFF)对兽用生物制品做出是否准予上市的行政审批。日本对兽用生物制品实施批签发管理开始于1994年,2008年起MAFF开始针对兽用生物制品施行产品基础种子批管理制度,且MAFF不再进行成品的批签发工作。MAFF对产品的基础种子进行注册批准后,授权各企业的质量管理部对产品进行日常批签发。日本的兽用生物制品批签发法律依据是《种子批质量控制体系综述》。
2.2 需要批签发的产品种类 所有兽用疫苗和诊断制品均需进行批签发。
2.3 兽用生物制品批签发流程 企业质检部门完成检验后,质管部门对样品进行抽样和留样。质管部经理检查产品的生产记录和自检报告,如果所有文件均符合要求,则由企业负责人对该产品进行批签发。批签发信息需提交到MAFF,供其掌握市场供应情况,流程见图2。
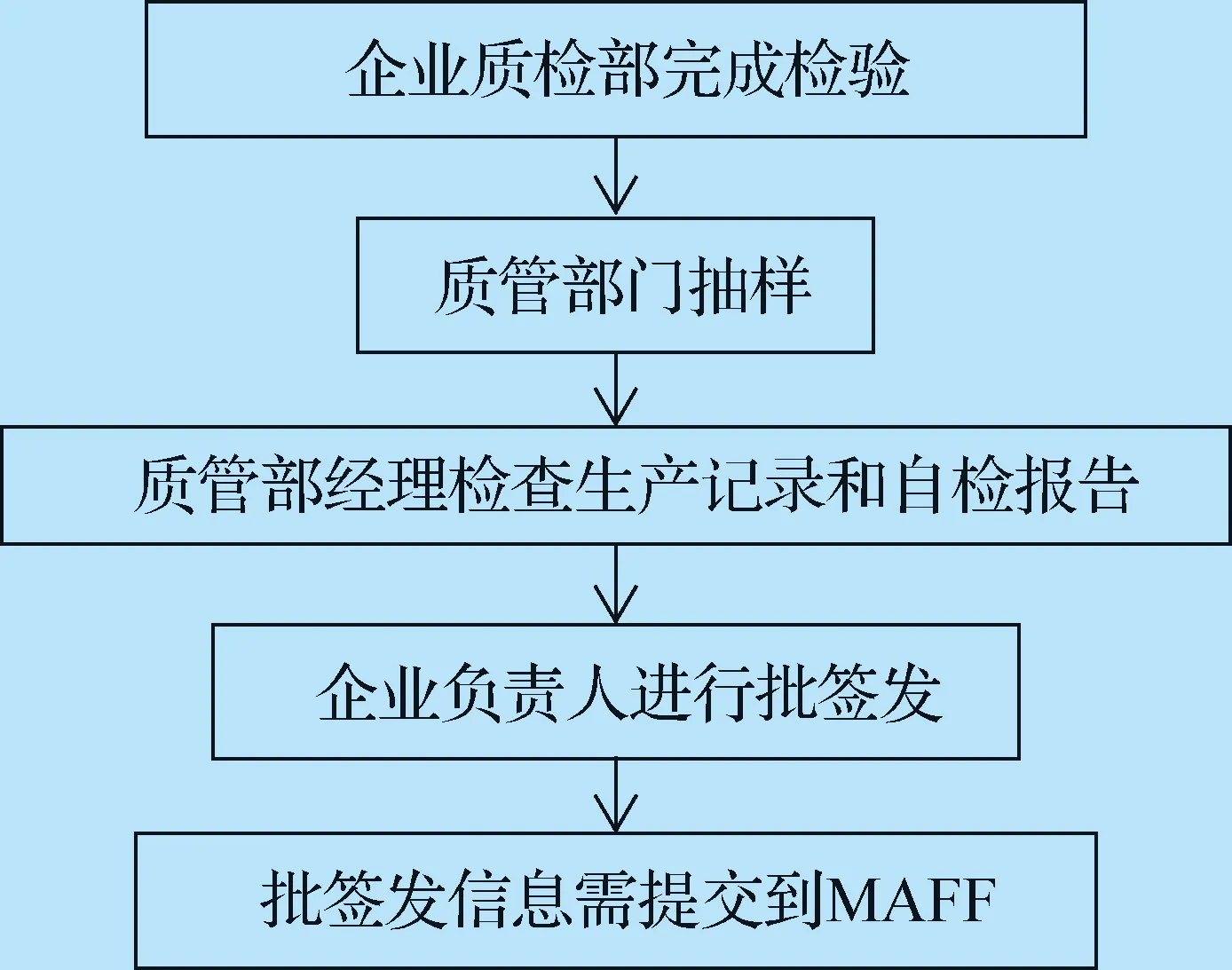
图2 日本兽用生物制品批签发流程
2.4 兽用生物制品批签发抽样检验 企业质管部门抽样应遵循随机抽样的原则。企业留样数量原则上应满足产品全检2次的用量,留样保留到有效期后12个月。一般产品无需MAFF进行检验。但日本的《国内传染病防控法典》中规定的重大动物疫病相关的疫苗需在批签发前将样品提交到MAFF指定的国家检验局进行检验,重大动物疫病类型见表1。

表1 日本《国内传染病防控法典》中规定的重大动物疫病类型
2.5 批签发核查和检查 日本兽用生物制品的批签发现场核查由MAFF负责开展。每5年结合GMP检查进行1次。
3 澳大利亚兽用生物制品批签发的管理情况
3.1 兽用生物制品批签发管理现状 澳大利亚的兽用生物制品批签发是指由澳大利亚农药兽药管理局(APVMA)对兽用生物制品做出是否准予上市的行政审批。澳大利亚对兽用生物制品实施批签发管理开始于1994年,自2001年起,APVMA不再进行日常批签发工作,而是授权各企业的GMP合规人负责本企业产品的日常批签发工作。澳大利亚的兽用生物制品批签发法律法规是《澳大利亚治疗药品管理法》。
3.2 需要批签发的产品种类 生产用抗原以及活疫苗、灭活疫苗、诊断试剂盒均需进行批签发。
3.3 兽用生物制品批签发流程 澳大利亚兽用生物制品批签发的整个过程均在企业内部完成,首先由企业质管部(QA)检查产品的生产记录和质检部门(QC)出具的产品自检报告,如果所有文件均符合要求,GMP合规人对该批产品进行批签发;如果产品自检不符合规定,则该批产品不予批签发;如果相关资料有其他不符合要求的情况,则由GMP合规人评估其对产品质量的影响,如果影响很小,则由合规人向APVMA提出产品批签发的书面申请,如果APVMA批准了该申请,则该批产品可予以签发,具体流程见图3。
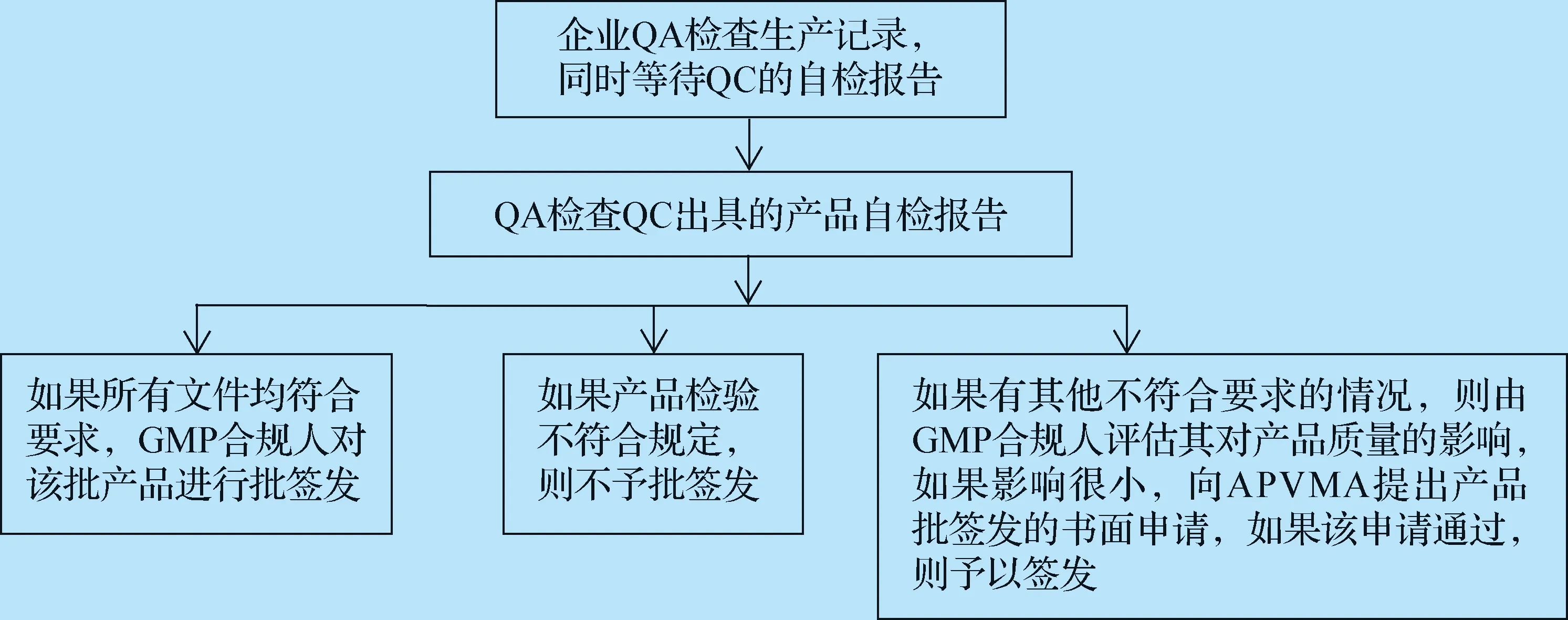
图3 澳大利亚兽用生物制品批签发流程
3.4 兽用生物制品批签发抽样检验 每批次产品在分装前、分装中或分装后,企业质检部门均会随机抽取样品并进行检验,如果产品的病毒滴度检验符合规定,则不需进行安全检验。质管部门同时负责样本的贴签和留样的保存工作。原则上按照样品全检2次所需的数量进行留样,样本保存至有效期后12个月。
3.5 批签发核查和检查 APVMA授权审核人每2年进行1次批签发现场核查,如果核查过程中发现不符合相关规定的情况,GMP合规人需承担相应的法律责任[2]。
4 三国批签发管理的共性、区别、特点及对我国管理工作的借鉴
4.1 三国的共性、区别和特点 在管理现状方面,韩、日、澳三国的共性是开展批签发管理的年代均较早,且有完善的法律法规支持批签发工作。韩国的管理模式与我国类似,由政府部门负责批签发,但韩国的兽用生物制品企业和产品数量都很少,且由于种类限制,年批签发量仅能达到我国批签发量的6%。澳大利亚和日本相似,政府部门不直接进行产品批签发管理,将批签发的管理权限下放到生产企业,日本由企业负责人进行批签发,澳大利亚则由GMP合规人进行批签发,GMP合规人一般职位是QA经理,如果合规人放行了不合格产品,需要承担相应的法律责任。日本的兽用生物制品批签发最大的特点是种子批制度,这种制度与世界其他国家的批签发管理方式均不相同,主要关注生产种子的质量,对成品不再进行批签发。在批签发的产品品种方面,韩国的产品种类较少,仅疫苗需要批签发,其他制品不需要进行批签发。日本和澳大利亚由于政府不直接进行批签发管理,因此批签发的种类与我国类似,基本涵盖了所有兽用生物制品。在批签发检验和留样方面,韩国的特点是免检制度,只要10批批签发产品合格就不再需要检验,此制度就意味着只要产品不出现质量问题,则多数产品上市前是不需要检验的。日本、澳大利亚两国的批签发检验即为企业自检的过程,日本的MAFF仅关注重大动物疫病。三国的批签发留样均在企业内部保存。在批签发现场核查方面,日本与我国相似,核查工作由政府部门负责,但日本每5年开展1次,而我国每年均开展现场核查。澳大利亚的现场核查委托第三方进行,一般政府部门不直接参与,韩国则不进行批签发现场核查。
4.2 对我国批签发管理工作的借鉴
4.2.1 建立兽用生物制品批签发法规体系 韩、日、澳三国虽然没有针对性的批签发法规,但在相应的法律规范中都对批签发相关内容进行了较为细致明确的规定。我国《兽药管理条例》中虽然有条款对产品上市销售前的批签发进行了阐述,但缺乏相应的细则。面临兽用生物制品发展速度快,批签发作为兽用生物制品上市销售的前置条件越来越受到关注的局面,亟待出台国家层面的针对性法律法规,为管理工作的科学化和规范化提供保证。
4.2.2 明确批签发管控重点,强化管理效能 韩、日、澳三国均为发达国家,对兽用生物制品开始实施批签发管理的年代也比较早,已经摸索出了一套适合自身特点的管理体系,且国民素质水平、企业的诚信度等均较高,因此管理部门更加信任企业,批签发管理更依赖于企业的自检自查。我国是全球兽用生物制品生产企业数量最多,年生产批数最多的国家,且我国目前处于较为敏感的社会转型期,在追求经济利益最大化的过程中,某些中小企业的诚信度就会有所缺失,因此我国不能完全参照发达国家的管理模式,在管理过程中不能盲目放手,而是要抓重点、强监管,主要关注重点产品的批签发。建议批签发类型参照韩国的模式,只对疫苗类产品进行批签发管理,一些体外诊断制品等可不纳入批签发管理范围,这样可将有限的监管精力用到刀刃上。同时应加大批签发检验和现场核查在管理过程中的作用,让批签发管理真正发挥实效[3]。
猜你喜欢
猪业科学(2022年10期)2022-11-03
上海计量测试(2022年2期)2022-08-30
吉林畜牧兽医(2022年7期)2022-07-20
猪业科学(2022年2期)2022-04-21
特种经济动植物(2021年4期)2021-12-12
民用飞机设计与研究(2020年4期)2021-01-21
林业科技(2020年3期)2021-01-21
中国动物保健(2018年6期)2018-12-28
中国动物保健(2018年9期)2018-12-14
声屏世界(2015年8期)2015-02-28
