
基于5993个核基因的被子植物系统发育关系研究
2020-03-02 07:46金鑫程书杨拓余慷段肖霞倪雪梅李世明张耕耘
广西植物 2020年1期
金鑫 程书 杨拓 余慷 段肖霞 倪雪梅 李世明 张耕耘

摘 要:系统发育关系的构建对被子植物分类及进化研究非常重要。长期以来,被子植物系统发育的研究,大多使用质体基因、线粒体基因或少数保守的单拷贝核基因。该研究从已注释基因组或转录组中搜集88种被子植物(包含58目)的核基因集;通过对其进行同源基因聚类及去旁系同源基因,获得了5 993个一对一的直系同源基因家族(即对于每个基因家族,每种植物最多一条序列,最少包含50个物种);使用截取各种不同数目基因集的DNA或氨基酸序列,采用串联法(concatenation)和溯祖法(coalescence),共构建了20棵进化树。比较这些进化树,虽然大部分结果支持APG IV中描述的被子植物主要支系之间的关系[(真双子叶植物,单子叶植物),木兰类植物],但真双子叶植物内部各目分支的演化关系与APG IV有一个很大的不同,即认为檀香目和石竹目是蔷薇类植物的姊妹群。基于这些进化树,估算了被子植物各目分支的分化时间,结果表明被子植物的起源时间为237.78百万年前(95%置信区间为202.6~278.08),与主流观点认为的225百万年至240百万年前一致。以上结果为构建进化树提供了一种可行性策略,这种方法允许使用基因数目更多而计算速度更快。
关键词:系统发育关系,被子植物,核基因,同源基因聚类,串联法,溯祖法,分化时间
中图分类号:Q949.4文献标识码:A
文章编号:1000-3142(2020)01-0044-16
Abstract:Construction of phylogeny is important for classification and research of angiosperms. For a long time,angiosperm phylogeny has been analysed using plastid genes,mitochondrial genes or a few conserved single-copy nuclear genes. We collected nuclear gene sets of 88 species of angiosperm (contains 58 orders) from annotated genomes or transcriptomes. By using a combined homology- and phylogeny tree-based approach,we obtained a total of 5 993 one-to-one ortholog groups (one sequence of each species for each ortholog group),each of which was represented by at least 50 species. Then,a total of 20 species trees were reconstructed using methods with different combinations of reconstruction (concatenation-based and coalescence-based) and sequence type (nucleotide or amino acid) for gene data sets with different gene occupancy values. Most of the resulting topologies support the relationships of the major clades of angiosperm as described in APG IV,but present different deep relationships among major clades in eudicots phylogeny such as the placement of Santalales and Caryophyllales as sisters to Rosids. We estimate the divergence times of the major clades of angiosperm and conclude that the origin of angiosperm is about 237.78 million years ago(95% confidence interval is 202.6-278.08),which is in accordance with the previously accepted 225 million years to 240 million years ago. This study provides an efficient strategy for building phylogenetic trees using thousands of genes with ultrafast calculation.
Key words:phylogeny,angiosperms,nuclear genes,ortholog inference,concatenation,coalescence,divergence time
系统发育树的正确构建对植物分类及进化研究非常重要。进化树构建的准确度主要受以下因素的影響。其一,所使用的数据集的种类及大小。不仅使用形态性状数据、质体基因、线粒体基因及核基因序列建立的进化树不一样(Endress & Doyle,2009; Soltis et al.,2011; Ruhfel et al.,2014; Zeng et al.,2014),使用全长核酸序列或仅使用基因密码子某个位点的核酸序列及氨基酸序列所构建的进化树也有所不同(Wickett et al.,2014);其二,构建树的方法及模型。方法有串联法(concatenation)和溯祖法(coalescence)。串联法是将所有基因串联作为一个整体,使用软件RAxML(Stamatakis,2014)或iqtree(Nguyen et al.,2015)构建系统发育树;溯祖法是先对每个基因建树,再使用软件ASTRAL(Zhang et al.,2017)建立所有基因树的共有树(Wickett et al.,2014)。而构建系统发育树使用的模型更是多种多样,如核酸模型GTR、HKY、JC、F81、K2P、K3P、K81uf等,蛋白质模型LG、Poisson、cpREV、mtREV、Dayhoff、mtMAM、JTT、WAG等(Nguyen et al.,2015)。
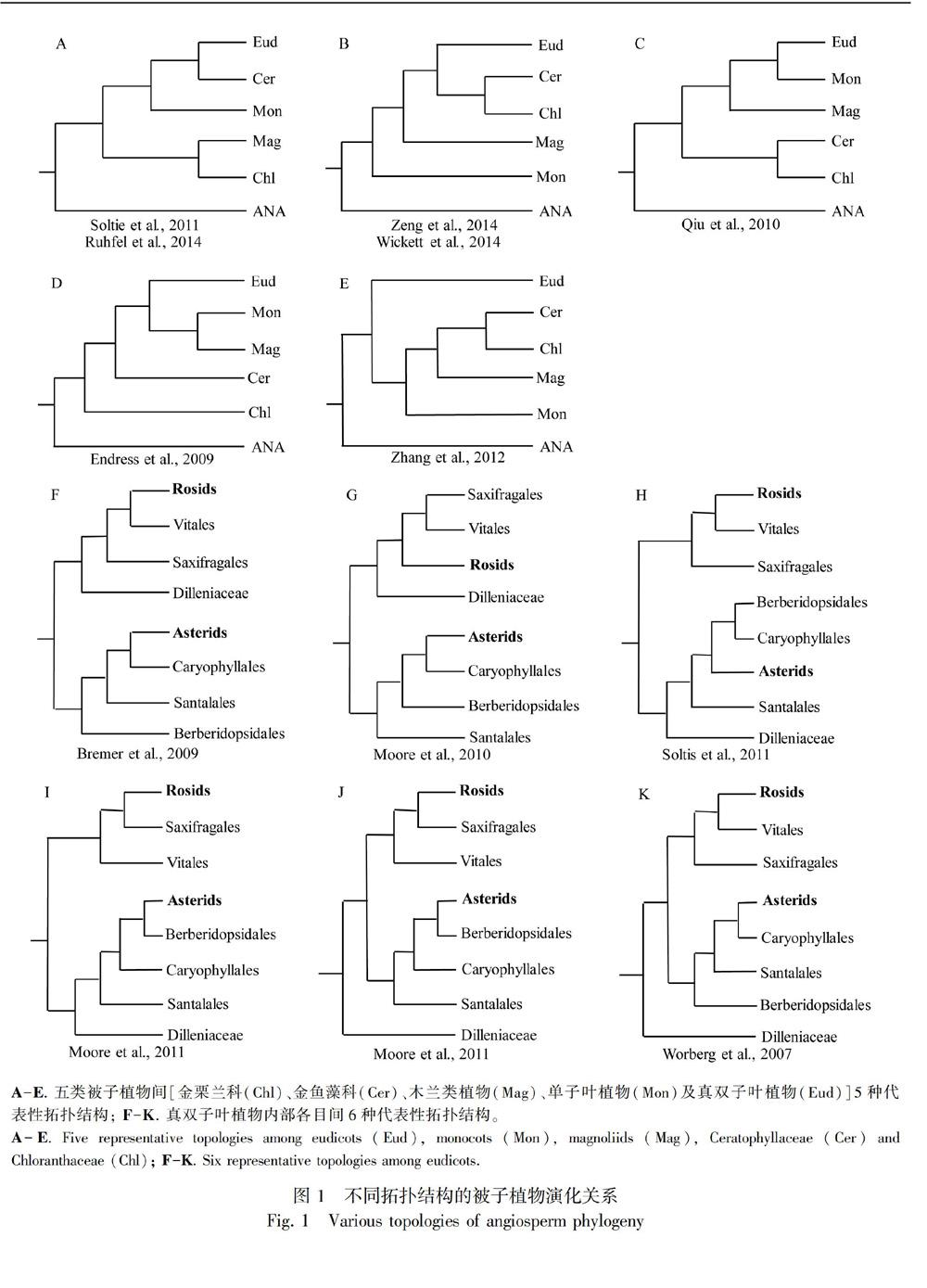
被子植物是植物界最高等且种类最多的一类,它们在地球上占据着绝对优势。现在已报道被子植物有352 000种(http://www.theplantlist.org/),属于416科和64目,各目之间的演化关系一直是研究的热点和争论的焦点。被子植物除了最基部的三个目:无油樟目(Amborellales)、睡莲目(Amborellales)和木兰藤目(Austrobaileyales),又称ANITA组,其余的(99.95%)可以分为五类:木兰类植物(magnoliids)、单子叶植物(monocots)、真双子叶植物(eudicots)、金栗兰科(Chloranthaceae)和金鱼藻科(Ceratophyllaceae)。这五类的系统演化拓扑关系一直存在争论,Zeng et al.(2014)总结了已经发表的五种主要的拓扑关系(图1:A-E),其中A是最主流的,也是APG IV(The Angiosperm Phylogeny Group,2016)的拓扑结构。Soltie et al.(2011)使用17个基因串联(包括质体基因、线粒体基因和核基因)为640种植物构建的系统发育进化树和Ruhfel et al.(2014)使用78个质体基因串联为360种植物构建的进化树,支持主流A拓扑结构。Wickett et al.(2014)使用674个核基因串联为92种植物构建的进化树和Zeng et al.(2014)使用59个核基因串联为61种植物构建的进化树,支持B拓扑结构。除此之外,Qiu et al.(2010)使用4个线粒体基因为380种植物构建的进化树,支持C拓扑结构;Endress & Doyle(2009)使用形态性状构建的进化树,支持D拓扑结构;Zhang et al.(2012)使用5个核基因为91种植物构建的进化树,支持E拓扑结构。
去掉金栗兰科和金鱼藻科后,单子叶植物、木兰类植物、真双子叶植物之间的系统发育关系有三种:(真双子叶植物,单子叶植物),木兰类植物;(真双子叶植物,木兰类植物),单子叶植物;(单子叶植物,木兰类植物),真双子叶植物。Lu et al.(2018)使用4个质体基因和1个线粒体基因分析了5 864种中国被子植物(几乎包括所有中国地区被子植物)的系统发育关系,其构建的进化树支持拓扑结构[(真双子叶植物,单子叶植物),木兰类植物]。Chen et al.(2019)发布了木兰类植物鹅掌楸(Liriodendron)基因组,使用其502个核基因及溯祖法为18种植物构建的进化树,同样支持拓扑结构[(真双子叶植物,单子叶植物),木兰类植物]。Chaw et al.(2019)发布了另一个木兰类植物牛樟(stout camphor tree)基因组,使用其211个核基因为13种植物构建的进化树,支持拓扑结构[(真双子叶植物,木兰类植物),单子叶植物]。Li et al.(2019)使用2 881种被子植物的质体基因组的80个基因重建了被子植物高分辨率的系統发育树,支持拓扑结构[(真双子叶植物,单子叶植物),木兰类植物]。从上述已有的研究中,我们发现,使用核基因串联法建立的进化树基本都支持拓扑结构[(真双子叶植物,木兰类植物),单子叶植物],使用核基因溯祖法、质体和线粒体基因建立的进化树基本都支持拓扑结构[(真双子叶植物,单子叶植物),木兰类植物]。
真双子叶植物内部各目的系统发育关系也存在争论(图1:F-K),真双子叶植物除了最基部的毛茛目(Ranunculales)、山龙眼目(Proteales)、昆栏树目(Trochodendrales)、黄杨目(Buxales)和洋二仙草目(Gunnerales),其余的可以分为两类:蔷薇类植物(Rosids)和菊类植物(Asterids)。这两类植物的基部有6个目的系统发育关系比较混乱,即五桠果目(Dilleniales)、虎耳草目(Saxifragales)、葡萄目(Vitales)、檀香目(Santalales)、智利藤目(Berberidopsidales)及石竹目(Caryophyllales)。Zeng et al.(2017)总结了已经发表的六种主要的拓扑关系(图1:F-K),其中K是APG IV中所认可的拓扑结构。Moore et al.(2010)使用83个质体基因为86种植物构建的进化树,支持“五桠果目是蔷薇类植物的姊妹群”;Soltie et al.(2011)使用17个基因串联(包括质体基因、线粒体基因和核基因)为640种植物构建的进化树和Moore et al.(2011)使用质体IR序列为87种植物构建的进化树,支持“五桠果目是菊类植物的姊妹群”;Worberg et al.(2007)等使用五个基因组区域序列为56种植物构建的进化树和Moore et al.(2011)使用质体IR序列为244种植物构建的进化树,及APG IV都支持“五桠果目是蔷薇类植物和菊类植物共同的姊妹群”。大部分研究都支持“葡萄目和虎耳草目是蔷薇类植物的姊妹群,智利藤目、檀香目和石竹目是菊类植物的姊妹群”(Worberg et al.,2007; Moore et al.,2010,2011; Yang et al.,2015);Zeng et al.(2017)使用504个核基因和溯祖法为100种植物构建的进化树,支持“檀香目和智利藤目是蔷薇类植物的姊妹群”。
被子植物的起源及进化一直是植物学界研究和争论的热点。在古生物学界,很长时期内,被子植物的最早化石记录都是白垩纪125百万年前,也是最早的真双子叶植物化石记录(Herendeen,1995)。Fu et al.(2018)发现了早侏罗纪地层(约175百万年前)中的“南京花”,其具有花萼、花瓣、雌蕊,有明显的杯托、下位子房上位花、树状的花柱,其种子/胚珠确实是被完全包裹着,子房壁将种子与外界完全隔绝,这都满足了被子植物判断标准。“南京花”的发现,将被子植物最早化石记录向前推进了约5 000万年,并填补了被子植物化石记录(125百万年前)与分子钟推算时间(225百万年至240百万年前)之间的“侏罗纪空缺”(Jurassic gap)(Li et al.,2019)。目前,大多数基于系统进化树的被子植物分化时间估计研究,都认为被子植物的起源为三叠纪225百万年至240百万年前(Magallon,2010; Smith et al.,2010; Zeng et al.,2014; Mandel,2019),这与起传粉作用的核心植食性鳞翅目昆虫的起源时间(约230百万年前)一致(Zeng et al.,2014; Li et al.,2019)。
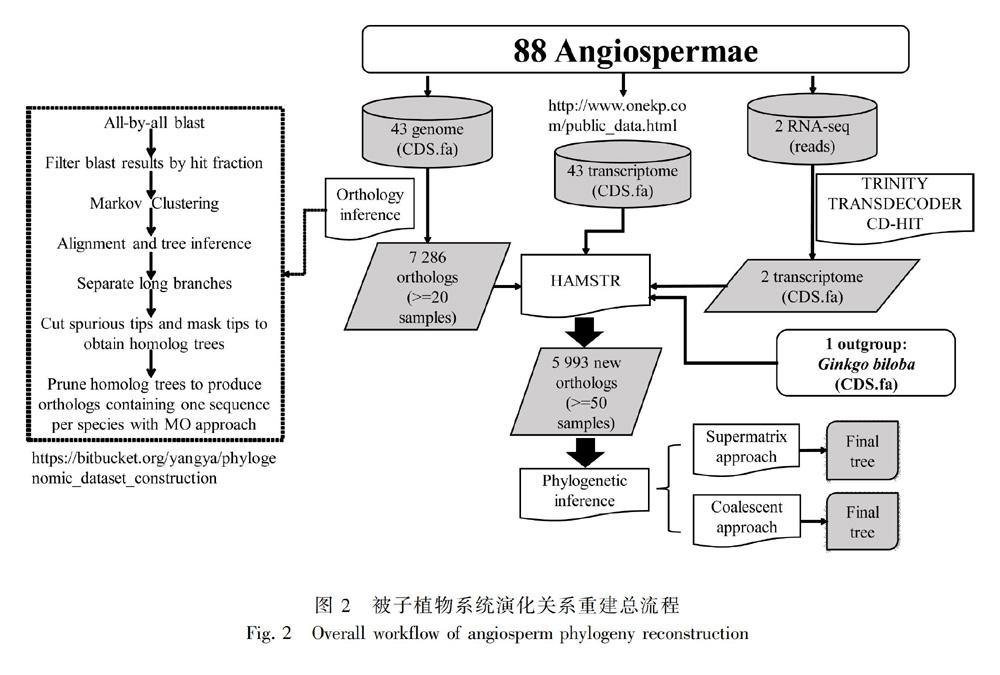
本研究使用超过5 000个核基因的核酸及蛋白序列,用两种进化树构建方法分析了88种被子植物的系统发育关系(包括87科58目),并对各进化分支的分化时间进行了估计(总流程如图2)。为了得到准确可靠的被子植物系统发育进化树,我们对5 000多个核基因进行了拆分,得到了包含不同基因数量的多个数据集,并对各个数据集进行系统发育树的构建,最后比较了所得到的20棵系统发育进化树之间的一致性。
1 材料与方法
1.1 材料
我们收集了1个裸子植物(Ginkgo biloba作为外类群)基因组、43个被子植物基因组(主要来自NCBI和PHYTOZOME数据库)、43个被子植物已拼接转录组(http://www.onekp.com/public_data.html )及2个被子植物RNA-seq数据(其中无叶莲Petrosavia sakurai是本研究测序的物种),A-E. 五类被子植物间[金栗兰科(Chl)、金鱼藻科(Cer)、木兰类植物(Mag)、单子叶植物(Mon)及真双子叶植物(Eud)]5种代表性拓扑结构; F-K. 真双子叶植物内部各目间6种代表性拓扑结构。
A-E. Five representative topologies among eudicots (Eud),monocots (Mon),magnoliids (Mag),Ceratophyllaceae (Cer) and Chloranthaceae (Chl); F-K. Six representative topologies among eudicots.其中被子植物共包含87科58目(表1)。
1.2 基于基因组序列的直系同源基因鉴定
我们使用Yang & Smith(2014)报道的方法,对43个植物基因组的基因集进行同源基因聚类分析。首先,使用软件BLASTN v2.6.0+ 对43个基因集CDS序列进行all-by-all blast,每条序列取最佳的1 000条比对结果,去掉比对长度小于1/3总长的序列,修剪未比对上的末端序列。然后,使用MCL软件(Van,2000)进行同源基因聚类(inflation value = 1.4),去除少于20个植物的基因家族,剩余基因家族使用MAFFT v7.310软件(Katoh & Standley,2013)进行多序列比对(maximum iterative refinement cycles = 1 000),使用PHYUTILITY v2.2.6软件(Smith & Dunn,2008)修剪缺失率大于90%的位点,使用软件RAXML v8.2.11(Stamatakis,2014)对修剪后的多序列比对数据估算系统进化树(model = GTRCAT)。最后修剪掉进化树上的所有旁系同源基因枝,修剪枝长大于0.6的枝、比姐妹枝长十倍的末端枝,单源且全部同样品的枝只保留一个,修剪枝长比预期碱基替换率大0.3倍的内部枝,再使用MO方法(Yang & Smith,2014)去除所有剩余的旁系同源枝,获得one-to-one同源基因家族(即每个样品最多一条序列),只保留大于20个样品的基因家族。
1.3 转錄组及外类群数据处理
我们对两个来自两个科(无叶莲科Petrosavia sakurai和鸦跖草科Cyanotis arachnoidea)的RNA-seq数据从头拼接。首先使用Trimmomatic v0.38软件(Bolger et al.,2014)过滤原始reads数据(参数HEADCROP:15 LEADING:20 TRAILING:20 SLIDINGWINDOW:5:20 MINLEN:50 AVGQUAL:20),再使用Trinity v2.6.6软件(Grabherr et al.,2011)拼接(min contig length=150 bp),最后使用TransDecoder v5.5.0(https://github.com/TransDecoder/TransDecoder/releases/tag/TransDecoder-v5.5.0 )进行CDS和蛋白质序列预测(参考数据库为Swissprot和Pfam-A)。将得到的这两个物种的基因集、从onekp数据库下载得到的43种被子植物的基因集和1个裸子植物(Ginkgo biloba)的基因集,使用HaMStR v13.2.6软件(Ebersberger et al.,2009)合并到利用基因组数据得到的同源基因家族中,最终只保留大于50个样品的基因家族。
1.4 系统发育进化树构建
我们采用两种方法串联法 (concatenation)和溯祖法(coalescence),并分别使用CDS序列和氨基酸序列构建进化树。无论是CDS序列还是蛋白质序列,都使用PRANK v.170427软件(http://wasabiapp.org/software/prank/)进行多序列比对,使用PHYUTILITY v2.2.6软件(Smith & Dunn,2008)修剪缺失率大于70%的位点,其中CDS序列需去除长度小于300个碱基的序列,蛋白质序列需去除长度小于100个氨基酸的序列。
溯祖法,先对每个基因使用RAxML v8.2.11软件(默认参数)(Stamatakis,2014)画树,再使用ASTRAL v5.5.9软件(Zhang et al.,2017)处理所有基因树,得到共有树,参数设置“-t 1--gene-only”以获得bootstrap值和基因支持率,枝长使用iqtree v1.5.5软件(Nguyen et al.,2015)获得。
串联法,先使用PartitionFinder v2.1.1软件(Lanfear et al.,2009)对串联序列进行分区和进化模型检测,从而设置较合理的分区和为每个分区选择合理的进化模型。对CDS序列检测下列的四个分区策略(表2):no partitioning,partitioning by each codon position (three partitions),partitioning by gene和partitioning by each codon position within each gene。对蛋白质序列检测下列两个分区策略:no partitioning和 partitioning by gene。参数设置如下:branch lengths = linked;model_selection = aicc;search = user;models = GTR,GTR+G,GTR+I+G(CDS序列)或者models = LG+G,LG+I+G,WAG+G,WAG+I+G(蛋白质序列)。再使用iqtree v1.5.5软件画树[1000 ultrafast bootstrap replicates(Von Haeseler et al.,2013),-spp设置最优分区策略],基因支持率使用ASTRAL v5.5.9软件(-t 1)获得。最后使用软件Evolview v2(He et al.,2016)对获得的所有进化树进行美化。
A. 每个同源基因家族含有的基因数目; B. 每个样品含有的同源基因家族数目。
A. Gene number of each orthologous gene family; B. Number of orthologous genes for each sample.
1.5 分化时间估计
我们使用PAML v4.9软件包 (Yang,2007)的MCMCTREE程序进行分化时间估计,输入拓扑结构为综合20棵进化树的最佳拓扑结构(即使用742个基因的CDS序列串联法获得的拓扑结构),输入序列为742个基因的CDS序列。我们先对每个基因都分别估计分化时间,再综合742个基因的分析结果(即每个节点取所有基因的平均值)获得最终的分化时间树。拓扑结构的枝长使用JONES+gamma碱基替换模型获得;rgene gamma设定为G(1,4.5);sigma2 gamma设定为G(1,4.5);clock设定为3;Markov chain Monte Carlo(MCMC)设定为burnin=50 000,sampfreq=100,nsample=10 000。对每个基因,都是分别运行两次独立的MCMC(即不同的random seeds),使用Tracer v1.7软件(https://github.com/beast-dev/tracer/releases/tag/v1.7.1 )观察运行结果是否稳定和收敛,所有节点及参数的effective sample size是否大于200。九个化石校准设定如下:银杏分化时间为290百万年至310百万年前(Gao et al.,1989);单子叶植物和真双子叶植物分化时间为130百万年至200百万年前(Kumar et al.,2017);真双子叶植物共同祖先(即最早的双子叶植物化石记录)为125 百万年前(Herendeen,1995; Zeng et al.,2014);山龙眼目(Proteales)的共同祖先为108.8百万年前(Crane et al.,1996);葡萄目(Vitales)与其余蔷薇类植物间分化时间为105百万年至115百万年前(Fawcett et al.,2009; Kumar et al.,2017);Arabidopsis thaliana与Populus trichocarpa间分化时间为97百万年至109百万年前(Kumar et al.,2017);豆目(Fabales)与壳斗目(Fagales)间分化时间为93.5百万年前(Friis et al.,1996);山茱萸目(Cornales)共同祖先为85.8百万年前(Takahashi et al.,2002);唇形目(Lamiales)共同祖先為44.3百万年前(Call et al.,1992)。
2 结果与分析
2.1 直系同源基因鉴定
我们对44个植物基因组基因集和45个已拼接转录组CDS序列进行同源基因聚类,并使用Yang & Smith(2014)报道的方法,去除所有旁系同源基因,最终获得大于50个样品的one-to-one 基因家族(即每个样品最多一条序列)共5 993个(图3:A),各种植物的基因覆盖率从33.57%到97.85%,平均为80.40%(图3:B)。
2.2 系统发育进化树构建
我们采用串联和溯祖法共构建了20棵进化树,并比较它们之间的不同(图4),以评估树的稳定性。CDS序列和蛋白质序列,都分别使用五个数据集,总共构建20棵树(5棵CDS串联法树,5棵CDS溯祖法树,5棵AA串联法树和5棵AA溯祖法树)。这5个数据集分别包含5 928个orthologs(≥50 samples)、3 384个orthologs(≥70 samples)、1 791个orthologs(≥80 samples)、742个orthologs(≥85 samples)及42个orthologs(≥89 samples)。
这20棵进化树主要是为了进一步确定图1中五类被子植物间演化关系和真双子叶植物内部各目间系统发育关系。这些进化树中的大多数,是与使用742个基因CDS序列(共4 069 848位点)串联方法建立的进化树高度一致的(图5)(使用3 384个基因AA序列建立的进化树,和使用1 791个基因AA序列建立的进化树,也是相同的最佳拓扑结构)。
2.2.1 木兰类植物、单子叶植物及双子叶植物间演化关系 无论核酸序列还是蛋白质序列,使用串联法和溯祖法建立的进化树基本都支持拓扑结构[(真双子叶植物,单子叶植物),木兰类植物](图4)。
2.2.2 金栗兰科与金鱼藻科 我们的研究表示,金鱼藻科是真双子叶植物的姊妹群,这与前人的研究结果一致(图4)。但金栗兰科是所有被子植物(除ANITA外)的基底旁系群,这与APG IV认为的“金栗兰科是木兰类植物的姊妹群”是不同的。
2.2.3 双子叶植物内部各目的系统发育关系 我们的研究认为,五桠果科是蔷薇类植物和菊类植物共同的姊妹群,虎耳草目是蔷薇类植物的姊妹群,这都与APG IV一致(图4)。
APG IV认为“檀香目和石竹目是菊类植物的姊妹群”,而我们的研究否定了这一结论:20棵进化树中,所有结果都支持“石竹目是蔷薇类植物的姊妹群”;大部分支持“檀香目是蔷薇类植物的姊妹群”,这与Zeng et al.( 2017)的研究结果一致;少部分支持“檀香目是蔷薇类植物和菊类植物共同的姊妹群”(图4)。
APG IV认为“智利藤目是菊类植物的姊妹群”,而我们的研究只有少部分支持这一结论。使用蛋白质序列建立的进化树,无论串联还是溯祖法,都支持“智利藤目是蔷薇类植物和菊类植物共同的姊妹群”。使用核酸序列建立的进化树,随着基因数目的增多,逐渐转变为支持“智利藤目是菊类植物的姊妹群”,与APG IV一致(图4)。
2.3 分化时间估计
基于742个基因CDS序列串联方法建立的进化树,我们估计了被子植物的分化时间(图6)。我们认为被子植物的起源时间为237.78百万年前(95%置信区间为202.6~278.08),与主流观点认为的225百万年至240百万年前一致 (Magallon,2010; Smith et al.,2010; Zeng et al.,2014)。木兰类植物与单子叶植物和真双子叶植物的分化时间约为166.11百万年前;五桠果科与蔷薇类和菊类植物的分化时间约为124.23百万年前;蔷薇类植物与菊类植物的分化时间约为116.98百万年前;唇形类植物(Lmiids)与桔梗类植物(Campanulids)的分化时间约为102.37百万年前。
3 讨论与结论
长期以来,被子植物的系统发育关系重建,都是使用质体基因、线粒体基因或少数保守的单拷贝核基因。Yang & Smith(2014)报道了一种基于系统进化树的同源基因聚类及去旁系同源基因的方法,我们使用此种方法对收集的88种植物核基因集进行聚类,共获得了多达5 993个one-to-one基因家族,并从这个数据集里面截取各种大小的数据进行进化树重建,以测定进化树的稳定性。
获得比以前更多的核基因家族后,制约系统演化关系构建的另一个因素就是大量的计算资源和计算时间。构建系统进化树时,一般需要设置bootstrap值(100~1 000)迭代,此步骤非常耗费计算时间。Nguyen et al.(2015)发表的软件iqtree,采用ultrafast bootstrap approximation(UFBoot)方法获得bootstrap值(Von Haeseler et al.,2013),比RAxML软件的传统方法,计算速度快10~40倍,并且获得的bootstrap值更精确。
我们使用多达5 993个one-to-one基因家族构建的进化树,与APG IV报道的主要差异为檀香目和石竹目在系统发育树中的位置,本研究认为“檀香目和石竹目是蔷薇类植物的姊妹群”,而APG IV认为“檀香目和石竹目是菊类植物的姊妹群”。可能原因有以下两个:一是基因数目的增多;二是本研究所选88个植物只有一半使用的基因组序列,另一半为转录组序列,而转录组序列一般存在大量的基因缺失(即未表达基因较多)。
A. Bootstrap值; B. 基因支持率。红色表示支持,蓝色表示拒绝,红色星星表示支持率最高的拓扑结构。
A. Bootstrap value; B. Gene trees support ratio. Red represents support,blue represents rejection,the topology labeled with a red star represents the most support.枝上斜线左边数字为bootstrap值,右边数字为基因支持率。
灰色条纹为分化时间的95%置信区间,九个化石校准时间为(1)银杏分化时间为290百万年至310百万年前;(2)单子叶植物和真双子叶植物分化时间为130百万年至200百万年前;(3)真双子叶植物共同祖先(即最早的双子叶植物化石记录)为125 百万年前;(4)山龙眼目(Proteales)的共同祖先为108.8百万年前;(5)山茱萸目(Cornales)共同祖先为85.8百万年前;(6)唇形目(Lamiales)共同祖先为44.3百万年前;(7)葡萄目(Vitales)与其余蔷薇类植物间分化时间为105百万年至115百万年前;(8)豆目(Fabales)与壳斗目(Fagales)间分化时间为93.5百万年前;(9)Arabidopsis thaliana与Populus trichocarpa间分化时间为97百万年至109百万年前。
Grey bars are 95% confidence intervals,nine fossil calibration points are as follows:(1) The divergence time of Ginkgo biloba is 290 million years to 310 million years ago; (2) The divergence time of eudicots and monocots is 130 million years to 200 million years ago; (3) The divergence time of eudicots is 125 million years ago; (4) The divergence time of Proteales is 108.8 million years ago; (5) The divergence time of Cornales is 85.8 million years ago; (6) The divergence time of Lamiales is 44.3 million years ago; (7) The divergence time of Vitales from Rosids is 105 million years to 115 million years ago; (8) The divergence time of Fabales and Fagales is 93.5 million years ago; (9) The divergence time of Arabidopsis thaliana and Populus trichocarpa is 97 million years to 109 million years ago.總的来说,本研究不仅进一步确定了被子植物各目间系统发育关系,而且为“使用更多的基因和计算速度更快的方法构建进化树”探讨了一种可行性策略:即使用Yang & Smith(2014)报道的同源基因聚类及去旁系同源基因方法,获得大量的one-to-one基因家族,再使用IQ-TREE(串联法)和ASTRAL(溯祖法)软件,能快速精确的计算出进化树。随着更多植物基因组的测序和基因聚类及系统发育关系构建方法的进一步优化,被子植物系统发育关系将越来越精确,例如进一步准确确定檀香目和石竹目在被子植物中与其他进化分支之间的关系。
參考文献:
BOLGERAM,LOHSE M,USADEL B,2014. Trimmomatic:A flexible trimmer for Illumina sequence data[J]. Bioinforma-tics,30(15):2114-2120.
CALL VB,DILCHER DL,1992. Investigations of angiosperms from the Eocene of southeastern North America:Samaras of Fraxinus wilcoxiana Berry[J]. Rev Palaeobot Palynol,74:249-266.
CHAW SM,LIU YC,WU YW,et al.,2019. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution[J]. Natl Plants,5(1):63-73.
CHEN JH,HAO ZD,GUANG XM,et al.,2019. Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation[J]. Nat Plants,5(1):18-25.
CRANE PR,HERENDEEN PS,1996. Cretaceous floras containing angiosperm flowers and fruits from eastern North America[J]. Rev Palaeobot Palynol,90:319-337.
EBERSBERGER I,STRAUSS S,VON HAESELER A,2009. HaMStR:Profile hidden markov model based search for orthologs in ESTs[J]. Bmc Evol Biol,9(1):157-157.
ENDRESS PK,DOYLE JA,2009. Reconstructing the ancestral angiosperm flower and its initial specializations[J]. Am J Bot,96(1):22-66.
FAWCETT JA,MAERE S,VAN DE PEER Y,2009. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event[J]. Proc Natl Acad Sci USA,106(14):5737-5742.
FRIIS EM,PEDERSEN KR,SCHNENBERGER J,2006. Normapolles plants:A prominent component of the Cretaceous rosid diversification[J]. Plant Syst Evol,260:107-140.
FU Q,DIEZ JB,POLE M,et al.,2018. An unexpected noncarpellate epigynous flower from the Jurassic of China[J]. Elife,7:e38827.
GAO Z,BARRY AT,1989. A review of fossil cycad megasporophylls,with new evidence of Crossozamia pomel and its associated leaves from the lower permian of Taiyuan,China[J]. REV Palaeobot Palynol,60(3-4):205-223.
GRABHERR MG,HAAS BJ,YASSOUR M,et al.,2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol,29(7):644-652.
HE ZL,ZHANG HK,GAO SH,et al.,2016. Evolview v2:An online visualization and management tool for customized and annotated phylogenetic trees[J]. Nucl Acid Res,44(W1):236-241.
HERENDEEN PS,1995. The enigma of angiosperm origins[J]. Earth-Sci Rev,39(1):253-254.
KATOH K,STANDLEY DM,2013. MAFFT multiple sequence alignment software version 7:Improvements in performance and usability[J]. Mol Biol Evol,30(4):772-780.
KUMAR S,STECHER G,SULESKI M,et al.,2017. TimeTree:A Resource for 598 timelines,timetrees,and divergence times[J]. Mol Biol Evol,34:1812-1819.
LANFEAR R,FRANDSEN PB,WRIGHT AM,et al.,2016. PartitionFinder 2:New methods for selecting partitioned models of evolution formolecular and morphological phylogenetic analyses[J]. Mol Biol Evol,34(3): 772-773.
LU LM,MAO LF,YANG T,et al.,2018. Evolutionary history of the angiosperm flora of China[J]. Nature,554(1):234-238.
LI HT,YI TS,GAO LM,et al.,2019. Origin of angiosperms and the puzzle of the Jurassic gap[J]. Nat Plants,5(1):461-470.
MAGALLON S,2010. Using fossils to break long branches in molecular dating:A comparison of relaxed clocks applied to the origin of angiosperms[J]. Syst Biol,59(4):384-399.
MOORE MJ,HASSAN N,GITZENDANNER MA,et al.,2011. Phylogenetic analysis of the plastid inverted repeat for 244 species:Insights into deeper-level angiosperm relationships from a long,slowly evolving sequence region[J]. Int J Plant Sci,172(4):541-558.
MOORE MJ,SOLTIS PS,BELL CD,et al.,2010. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots[J]. Proc Natl Acad Sci USA,107(10):4623-4628.
NGUYEN LT,SCHMIDT HA,VON HAESELER A,et al.,2015. IQ-TREE:A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies[J]. Mol Biol Evol,32(1):268-274.
QIU YL,LI LB,WANG B,et al.,2010. Angiosperm phylogeny inferred from sequences of four mitochondrial genes[J]. JSE,48(6):391-425.
RUHFEL BR,GITZENDANNER MA,SOLTIS PS,et al.,2014. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes[J]. Bmc Evol Biol,14(1):23.
SMITH SA,BEAULIEU JM,DONOGHUE MJ,2010. An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants[J]. Proc Natl Acad Sci USA,107(13): 5897-5902.
SMITH SA,DUNN CW,2008. Phyutility:A phyloinformatics tool for trees,alignments and molecular data[J]. Bioinformatics,24(5):715-716.
SOLTIS DE,SMITH SA,CELLINESE N,et al.,2011. Angiosperm phylogeny:17 genes,640taxa[J]. Am J Bot,98(4): 704-730.
STAMATAKIS A,2014. RAxML Version 8:A tool for phylogenetic analysis and post—analysis of large phylogenies[J]. Bioinformatics,30(9):1312-1313.
TAKAHASHI M,CRANE PR,MANCHESTER SR,2002. Hironoia fusiformis gen. et sp. nov.,a cornalean fruit from the Kamikitaba locality (Upper Cretaceous,Lower Coniacian) in northeastern Japan[J]. J Plant Res,115:463-473.
THE ANGIOSPERM PHYLOGENY GROUP,2016. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants:APGIV[J]. Bot J Linn Soc,181(1):1-20.
VANDS,2000. Graph Clustering by Flow Simulation[M]. University of Utrecht.
VONHAESELER A,MINH BQ,NGUYEN MAT,2013. Ultrafast approximation for phylogenetic bootstrap[J]. Mol Biol Evol,30(5):1188-1195.
WICKETT NJ,MIRARAB S,NGUYEN N,et al.,2014. Phylotranscriptomic analysis of the origin and early diversification of land plants[J]. Proc Natl Acad Sci USA,111(45): 4859-4868.
WORBERG A,QUANDT D,BARNISKE AM,et al.,2007. Phylogeny of basal eudicots:Insights from non-coding and rapidly evolving DNA[J]. Org Divers Evol,7(1):55-77.
YANG Z,2007. PAML 4:Phylogenetic analysis by maximum likelihood[J]. Mol Biol Evol,24:1586-1591.
YANG Y,MOORE MJ,BROCKINGTON SF,et al.,2015. Dissecting molecular evolution in the highly diverse plant clade caryophyllales using transcriptome sequencing[J]. Mol Biol Evol,32(8):2001-2014.
YANG Y,SMITH SA,2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes:Improving accuracy and matrix occupancy for phylogenomics[J]. Mol Biol Evol,31(11):3081-3092.
ZENG LP,ZHANG N,ZHANG Q,et al.,2017. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets[J]. New Phytol,214(3):1338-1354.
ZENG LP,ZHANG Q,SUN RR,et al.,2014. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times[J]. Nat Comm,5(1): 4956.
ZHANG C,SAYYARI E,MIRARAB S,2017. ASTRAL-III:Increased scalability and impacts of contracting low support branches[J]. RECOMB-CG,Springer,Cham:53-75.
ZHANG N,ZENG LP,SHAN HY,et al.,2012. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms[J]. New Phytol,195(4):923-937.
(責任编辑 周翠鸣)
