
中国儿童谷氨酸脱氢酶型先天性高胰岛素血症临床及遗传学特征分析
2020-03-02 08:26吕葛徐子迪惠培培曾俏刘敏闫洁吴玉筠桑艳梅
中华胰腺病杂志 2020年1期
吕葛 徐子迪 惠培培 曾俏 刘敏 闫洁 吴玉筠 桑艳梅
1国家儿童医学中心,首都医科大学附属北京儿童医院检验中心,北京 100045;2国家儿童医学中心,首都医科大学附属北京儿童医院内分泌遗传代谢中心,北京 100045
先天性高胰岛素血症(congenital hyperinsulinism, CHI)是一种遗传异质性疾病,由于胰岛素自律性分泌过多,血胰岛素浓度增加,从而导致出生后出现顽固性、持久性低血糖的表现,是婴幼儿和儿童时期持续性、复发性低血糖的重要原因。迄今已发现至少14种基因突变与CHI的遗传发病机制有关,表现为13种遗传学类型,不同类型CHI患儿的临床特征、治疗方案、治疗效果及预后不尽相同[1-4]。谷氨酸脱氢酶型先天性高胰岛素血症(glutamate dehydrogenase hyperinsulinism, GDH-HI)是CHI的第2种常见类型,由编码线粒体酶谷氨酸脱氢酶(glutamate dehydrogenase,GDH)的GLUD1基因功能增强性突变引起。本研究分析10例GDH-HI患儿的临床及遗传学特征,以期提高临床医师对该病的认识。
资料和方法
一、研究对象
选取2008年2月至2018年12月间北京儿童医院收治的10例临床初步诊断为GDH-HI患儿家系为研究对象。CHI诊断指标包括高胰岛素血症(胰岛素>2 mU/L)、低脂肪酸血症(游离脂肪酸<1.5 mmol/L)、低酮血症(β-羟丁酸<2.0 mmol/L)、1 mg静脉胰高血糖素试验反应血糖变化>0.3 g/L[5]。本研究经医院伦理委员会批准,所有患儿父母均签署知情同意书。
二、遗传学分析
留取患儿及父母的血液标本3 ml,采用BloodGen Midi Kit(CWBIO,中国)提取患者全基因组DNA,然后分别应用聚合酶链反应DNA(PCR-DNA)直接测序技术(病例1、3、4和5)和二代测序技术(其余6例病例)进行相关致病基因分析。
PCR-DNA直接测序:利用Premier5.0软件设计用于扩增GLUD1基因第6、7、10、11、12外显子区域的引物(表1)。PCR反应体积为50 μL,含5×GoTaq缓冲液10 μL、2.0 mmol/L Mg2+、0.2 mmol/L dNTP、正反向引物各1 μmol/L,1.25 U热启动Taq酶(美国Promega公司)。在Labnet热循环仪(美国Labnet公司)扩增,PCR反应条件:94℃ 5 min,94℃ 30 s、58~62℃ 30 s,72℃ 45 s,循环38次,最后72℃ 5 min。PCR扩增产物经琼脂糖凝胶电泳分离、回收片段,采用天根公司胶回收试剂盒纯化产物,由北京英俊公司行直接测序。

表1 GLUD1基因扩增引物序列
二代测序:参考OMIM数据库信息[1-4],将与低血糖相关的基因组外显子区通过捕获探针(SeqCap EZ MedExome Enrichment Kit 探针,瑞士罗氏公司)进行目标基因全外显子捕获,经Illumina hiseq XTen平台标准化上机测序。若经二代测序发现患儿携带GLUD1基因突变,则根据基因所验证位点序列设计引物,采用PCR方法进行扩增,引物序列及扩增条件同上,扩增产物纯化后上ABI 3730XL测序仪测序,采用DNASTAR软件进行基因序列分析和比对。
结 果
一、临床特征
10例患儿中男性8例,女性2例,出生体重为2.65~4.25 kg;9例为足月适龄儿,1例为巨大儿;发病年龄为出生后1 d~14个月,中位年龄为8个月。8例患儿首发症状为抽搐,2例首发症状为反应弱。患儿均无低血糖家族史。10例患儿血糖为1.40~2.60 mmol/L,均低于正常值;血胰岛素为3.54~45.02 mU/L,均高于正常值;9例伴有无症状性高氨血症(血氨正常值为18~72 μmol/L),1例患儿(病例7)血氨正常,为巨大儿。10例患儿确诊为CHI后,其中9例予以二氮嗪试验性治疗。二氮嗪起始剂量为5 mg·kg-1·d-1,每日2~3次口服,并根据患儿病情逐渐增加剂量,最大剂量为15 mg·kg-1·d-1。同时加用氢氯噻嗪利尿(1~2 mg·kg-1·d-1,分2~3次口服),防止水钠潴留,并加用10%氯化钾1~2 ml· kg-1·d-1,分3次口服,防止利尿剂引起的低钾血症。9例患儿均对二氮嗪治疗有效。另1例未进行治疗(表2)。
二、遗传学特征
10例患儿均发现携带GLUD1基因(NM_005271.5)突变,均为错义突变。病例1携带c.1388A>T突变,致p.N463I(外显子10),为新生突变;病例2携带c.1495C>A突变,致p.G499C(外显子12),为新生突变;病例3、4、7、9、10均携带c.965C>T突变,致p.R322H(外显子7),其中病例3为父系常染色体显性遗传,其余均为新生突变;病例5携带c.1493C>T突变,致p. S498L(外显子11),为新生突变;病例6携带c.1519G>A突变,致p.H507Y(外显子12),为新生突变;病例8携带c.1388A>G突变,致p.N463S(外显子10),为新生突变(表2)。
三、随访结果
经过长期随访,1例患儿确诊CHI后未曾试用二氮嗪治疗,通过口服葡萄糖维持正常血糖水平,在11岁左右病情自行缓解。曾用二氮嗪治疗的9例患儿中7例继续口服二氮嗪治疗,可维持正常血糖,其中2例同时患有癫痫;2例患儿失访(表2)。
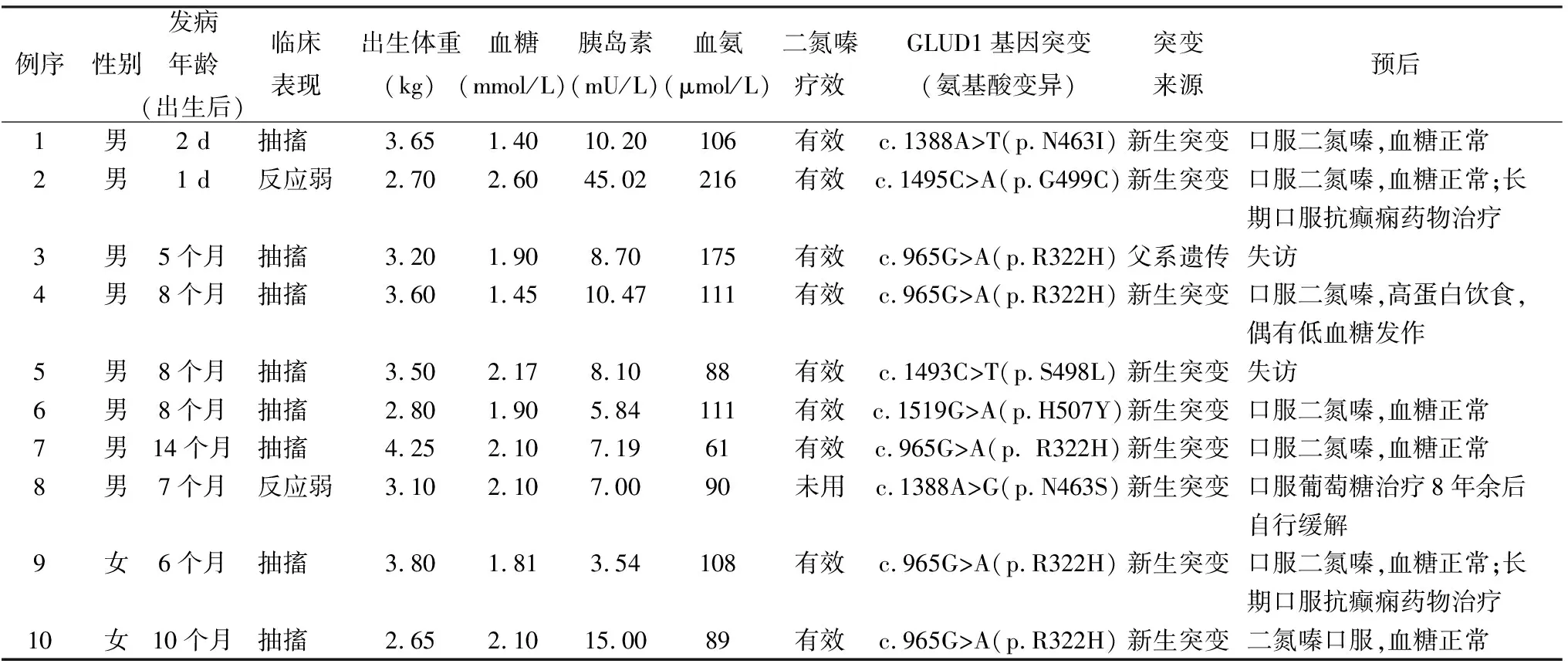
表2 10例谷氨酸脱氢酶型先天性高胰岛素血症患儿临床资料和预后情况
讨 论
GDH-HI是CHI的第2种常见类型,约占CHI的5%。1956年Cochrane等[6]首次报道了高蛋白饮食(尤其是亮氨酸)可导致低血糖,当时命名为“亮氨酸敏感性低血糖”。1998年Stanley等[7]首次描述了GDH-HI,也称为高胰岛素血症-高氨血症综合征(hyperinsulinism-hyperammonemia syndrome,HI-HA),是由线粒体基质酶中GDH的功能增强性突变引起的疾病。GDH由GLUD1基因编码, 是一种别构蛋白,为六聚体结构。成熟的GDH由两套三聚体组成,在每个三聚体中,酶的亚单位通过突出的触角样结构域结合在一起,使酶的各亚单位之间相互协调一致。GLUD1基因位于染色体10q23.3,含有13个外显子,编码505个氨基酸。
GDH-HI发病年龄跨度较大,患儿绝大多数在出生后4个月或以后,新生儿期发病者少见,也有部分为成年后才确诊。出生体重多正常。临床表现多为低血糖症状,如无明显诱因的抽搐、口唇青紫、口周发青、肌张力减弱、反应减弱、嗜睡等。患儿的低血糖症状可由空腹和高蛋白(尤其是亮氨酸)饮食诱发。本研究10例患儿的发病年龄为出生后1 d至14个月,9例患儿出生体重正常,与文献报道基本一致[8-9]。
随着研究的不断深入,迄今已发现了37种GLUD1基因杂合突变,目前仅报道过1例GDH-HI患儿同时携带母系遗传的移码突变(c.37delC)和新生突变c.1493C>T(p. S498L),其余突变类型集中在第6、7、10、11、12外显子区的错义突变[10]。突变位于外显子11、12者约占48%,位于外显子6、7者约占29%。本研究10例患儿共发现了6种GLUD1基因突变类型,其中携带GDH外显子7 p.R322H突变者占50%(5/10),携带外显子11、12突变者占30%(3/10),携带外显子10突变者占20%(2/10),提示GDH p.R322H可能是中国GDH-HI患儿的热点突变类型。本组患儿的突变分布比例与国外文献报道有所不同,考虑与样本量较小或人种差异有关。 GDH-HI的遗传方式为常染色体显性遗传或新生突变,其中常染色体显性遗传约占20%,新生突变约占80%[11]。 本研究10例患儿中9例为新生突变,1例为父系遗传的常染色体显性遗传,遗传方式与既往报道一致[2]。
GDH-HI除可引起患儿频发的低血糖,一般还伴有无症状性、持续性高氨血症(可达正常值上限的3~5倍)。患儿的高血氨值不会因低血糖发作、高蛋白饮食而升高,也不会因限制蛋白饮食或二氮嗪治疗而降低,但并非所有的GDH-HI患者均存在高氨血症[12]。产生高氨血症的机制主要包括:(1)GDH活性增强,导致谷氨酸产生的氨增加。(2)谷氨酸的消耗增多,减少N-乙酰谷氨酸的产生,而后者是氨解毒第一步所必需的变构活化剂。注射N-乙酰谷氨酸的类似物N-氨基甲酰谷氨酸可降低血氨,以此可证明该机制的存在[13]。本研究10例中9例表现出高氨血症,1例血氨值正常。2001年Santer等[14]曾报道2例携带GDH p.R322H突变的患儿,其血氨值均正常,此现象的发生可能是由于患儿为GLUD1基因突变的嵌合体(突变细胞与正常细胞并存),在肝脏组织中该基因突变缺失或突变率<50%。相关研究结果提示高氨血症并不具有基因特异性,但是需要进一步进行肝脏或胰腺活检来证明这种可能性[13]。
文献报道,46%~64%的GDH-HI患儿可伴有癫痫,他们的基因突变多发生在外显子6和7[13],携带GDH外显子10、11、12的患儿也可发生癫痫,癫痫最早可在新生儿时期出现[15]。本研究中2例患儿患有癫痫,病例2于低血糖发病1个月后(2月龄)确诊癫痫,病例9于低血糖发病3个月后(9月龄)确诊癫痫,突变类型分别为GDH p.R322H和p.G499C,分别位于GDH的第7和12外显子,这种不同突变位置与基因型-表现型的相关机制尚不明确。
因患儿胰腺β细胞的ATP敏感性钾通道是正常的,故绝大多数GDH-HI患儿对二氮嗪治疗有效[16-18],目前尚未发现有文献报道二氮嗪治疗无效的GDH-HI。本研究 10例中9例试用二氮嗪治疗,均有效,与上述文献报道一致。GDH-HI患儿建议给予低蛋白饮食作为辅助治疗,每餐亮氨酸≤200 mg可有助于降低二氮嗪的用量[19]。
本研究8例经过长期随访,其中7例患儿均继续服用二氮嗪进行治疗,可维持空腹血糖正常。长期应用二氮嗪过程中,除部分患儿有多毛表现外,未见其他药物不良反应。研究资料显示,随着CHI患儿的年龄增长及其对胰岛素需要量的增加,其低血糖症状可有所减轻,部分患儿可自愈。有研究对101例CHI患儿随访发现,约48%的CHI患儿低血糖可自行缓解,且自行缓解的时间跨度较长(3个月至8岁左右);在未检测出已知致病基因突变的患儿中,约65%患儿可自行缓解。而自行缓解的患儿中约15%是ATP敏感性钾离子通道型先天性高胰岛素血症(由ABCC8或KCNJ11基因突变导致),另有4%的患儿携带GLUD1、GCK或HNF4α基因突变[20]。本组1例患儿未试用二氮嗪,间断口服葡萄糖溶液来维持血糖正常,于11岁时低血糖症状自行缓解,提示携带GDH-HI突变的患儿存在自行缓解的可能性。
总之,中国儿童GDH-HI的临床特征和遗传学特征与其他人种相比差异并无统计学意义,但GDH p.R322H突变为中国儿童GDH-HI的热点突变类型。GDH-HI患儿多对二氮嗪治疗有效,部分患儿可出现癫痫。随着病程的进展,少数患儿存在自行缓解的可能。值得一提的是,少数患儿血氨可正常,临床须注意避免漏诊。细致的致病基因分析对患儿的遗传学分型、诊疗方案的确立、随访、预后的判断和遗传咨询等均有重要意义。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国饲料(2021年17期)2021-11-02
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
肿瘤预防与治疗(2019年6期)2019-07-30
分析化学(2019年3期)2019-03-30
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
复旦学报(医学版)(2016年6期)2016-12-20
医学研究杂志(2015年12期)2015-06-10
