
Triterpenoidsfrom Ainsliaea latifolia and Their Cyclooxyenase-2(COX-2)Inhibitory Activities
2020-03-02 04:46WenLinYuanXueYunDongZhengRuiHuangSiJiaXiaoJiYeXinHuiTianHuiLiangLiYunHengShenWeiDongZhang
Wen-Lin Yuan ·Xue-Yun Dong ,4·Zheng-Rui Huang ·Si-Jia Xiao ·Ji Ye ·Xin-Hui Tian ·Hui-Liang Li ·Yun-Heng Shen ·Wei-Dong Zhang ,
Abstract
KeywordsAinsliaea latifolia ·Triterpenoids·COX-2·Cucurbitane
1 Introduction
The genusAinsliaea(Compositae),a medicinally important genus in traditional Chinese medicine,comprises about 70 species worldwide,in which mostAinsliaeaplants are distributed in Southeast Asia.Previous investigations have reported the presence of sesquiterpenoids,sesquiterpene lactone dimers,triterpenoids,steroids and flavonoids inAinsliaeaspecies [1-3].Some of them exhibited diverse biological activities,including cytotoxic,antiviral,antibacterial and anti-inflammatory activities [4-6].
Ainsliaea latifoliagrows mainly in the southwest of China and has long been used as a folk medicine for the treatment of rhumatism,traumatic injuries,edema,stomachache,and anorexia [7].InAinsliaeaspecies,sesquiterpenoids are usually considered as characteristic chemical constituents.However,in our study of the chemical constituents fromA.latifolia,eight new triterpenoids (1-8) and one known triterpenoid (9) were isolated and identified from the whole plants ofA.latifolia.Herein,we described the isolation and structural elucidation of compounds 1-8,as well as their inhibition against cyclooxyenase-2 (COX-2).
2 Results and Discussion
The CHCl3-soluble of the EtOH-H2O (80:20,v/v) extract ofA.latifoliawas purified by repeated column chromatography (CC) over silica gel,Sephadex LH-20,and semi-preparative HPLC to yield eight new and one known compounds.By comparison of their NMR and MS data with the published references,the known compound 9 was then identified as one triterpenoid cucurbita-5,23-diene-3β,25-diol (9) [8].The structures of eight new triterpenoids were determined by analysis of HRESIMS and NMR spectroscopic data (Fig.1).
Compound 1 was isolated as white solid.Its molecular formula (C30H50O3),ascertained via high resolution ESI-MS analysis,indicated six degrees of unsaturation.The1H NMR spectrum of 1 (Table 1) exhibited signals for three olefinic protons atδH5.59 (2H),5.42 (1H,m),two oxygenated methine groups atδH3.83 (1H,d,J= 7.1 Hz),3.47 (1H,brt,J= 2.5 Hz),eight methyl groups (δH1.20,1.14,1.13,1.02,1.00,0.92,0.87,0.81).The13C NMR spectrum revealed the presence of thirty carbon signals including four olefinic carbons atδC141.2,141.3,125.7 and 121.4,three oxygenated carbons atδC79.7,76.6 and 72.9,and eight methyl carbons atδC28.0,27.2,26.3,25.4,23.7,20.4,17.8 and 15.7.The other carbon signals were assigned to seven methylenes,four methines,and four quaternary carbons.A comparison of these carbon resonances with those of the related cucurbitane-type triterpenoids suggested that 1 possessed the same cucurbitane skeleton,and the differences between the spectroscopic data of 1 and those of known compound 9 wereprimarily the observation of an oxymethine and the absence of a methylene.In the 1 H-1H COSY spectrum of 1,two mutual coupling olefinic protons exhibited the correlations with H-20 and the oxygenated methine proton atδH3.83 (Fig.2),respectively,ascribing a double bond to C-22 and C-23 positions.The HMBC correlation (Fig.2) of CH3-21 with the olefinic carbon atδC141.3 confirmed the above deduction.Also,the observation of HMBC correlations from CH3-26 and CH3-27 to C-24 (δC79.7) and the oxygenated quaternary carbon atδC72.9 supported the hydroxyl substituents at C-24 and C-25 positions.The absolute configuration of C-24 in 1 was assigned using the Snatzke’s method [9,10].Metal complex of compound 1 in DMSO gave a significant induced CD spectrum (ICD) (Fig.4),in which the positive cotton effect observed at 315 nm permitted the assignment of a 24Sconfiguration for 1.The relative configurations of other stereocenters of 1 were established to be identical to those of known compound 9 due to NOESY experiment (Fig.3).Thus,the structure of compound 1 was identified as cucurbita-5,22-diene-3β,24S,25-triol.
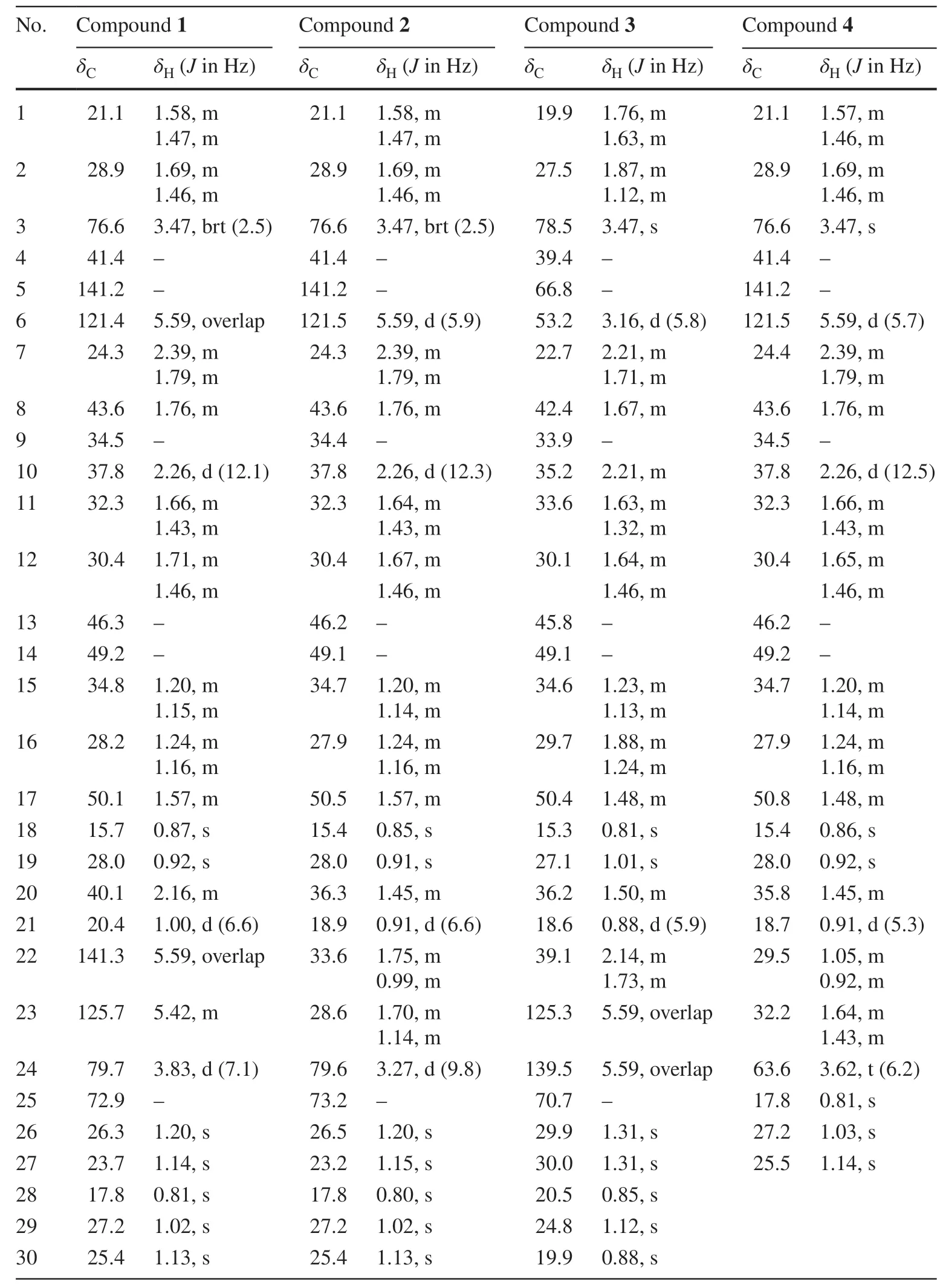
Table 1 1H (500 MHz) and 13 C (125 MHz) NMR spectroscopic data of compounds 1-4 in CDCl 3
Compound 2 was obtained as white solid and assigned a molecular formula of C30H52O3(HRESIMSm/z495.3622 [M+Cl]-,calcd for 495.3610),with two hydrogen atoms more than that of 1 (493.3447 [M+Cl]-).The1H and13C NMR spectra (Table 1) of 2 were very similar to 1,except that two olefinic protons of 1 were replaced by two methylenes in 2.Therefore,the structure of 2 was determined to be a hydrogenated derivative of 1 at C-22/C-23 double bond.The assignment was confirmed by the1H-1H COSY correlations of CH3-21/H-20/CH2-22/CH2-23/H-24 and key HMBC correlations of the oxygenated methine proton atδH3.31 (H-24) with C-22 and C-23,and of CH3-26 and CH3-27 with C-24 (δC79.6).Similarly,the absolute configuration of C-24 in 2 was confirmed using the Snatzke’s method [9,10].The positive Cotton effect observed at 310 nm (Fig.4) permitted the assignment of a 24Sconfiguration for 2.Thus,the structure of compound 2 was identified as cucurbita-5-ene-3β,24S,25-triol.
Compound 3 was isolated as white solid.Its molecular formula (C30H50O3),ascertained via high resolution ESI-MS analysis,indicated six degrees of unsaturation.Detailed analysis of the NMR (Table 1) and MS spectra led to the conclusion that the only difference between 3 and known compound 9 was that there is an epoxide group between C-5 (δC66.8,s) and C-6 (δC53.2,d) in 3 instead of a double bond between C-5 (δC141.2,s) and C-6 (δC121.4,d) in 9.The epoxide group was elucidated by HMBC correlations of H-1,H-3,H-7,CH3-29 and CH3-30 with C-5,and of H-8 and H-10 with C-6,as well as the1H-1H COSY correlations of H-6/H-7.The NOESY correlations of H-6/CH3-29 indicated the epoxy ring of 3 was inβ-orientation.Thus,the structure of compound 3 was identified as cucurbita-5β,6βepoxy-23-ene-3β,25-diol.
Compound 4 was obtained as white solid and assigned a molecular formula of C27H46O2,(HRESIMSm/z403.3594 [M+H]+,calcd for 403.3571),indicating five degrees of unsaturation.In the1H NMR spectrum (Table 1),the signals of five tertiary methyl groups (δH1.14,1.03,0.92,0.86,0.81) and one secondary methyl group (δH0.91,3H,d,J= 5.3 Hz) were observed.The13C NMR spectrum of 4 showed signals for 27 carbons due to six methyl groups,two olefinic carbons,ten methylenes (including an oxygenated one),five methines (including an oxygenated one),and four quaternary carbons.Detailed comparison of the13C NMR spectrum of 4 with that of 2 displayed similarities in rings A-D,except for the absence of the signals for C-25,26,27.These evidences revealed that compound 4 is a rare 25,26,27-trinorcucurbitane triterpenoid.This can be confirmed via the1H-1H COSY correlations of H3-21/H-20/H2-22/H2-23/H2-24.Thus,the structure of compound 4 was identified as 25,26,27-trinorcucurbita-5-ene-3β,24-diol.
Compound 5 was isolated as white solid.Its molecular formula (C27H44O3),ascertained via high resolution ESI-MS analysis,indicated six degrees of unsaturation.Analysis of the1H and13C NMR spectroscopic data of 5 (Table 2) indicated a structural similarity with compound 4,except that compound 5 has a carboxyl (δC178.8,C-24) instead of hydroxyl methyl signals in 4.The deduction was confirmed via the HMBC correlations from H-22,H-23 to the carboxyl carbon (C-24).The relative configurations of 5 were evidenced to be identical to those of 4 by analysis of NOESY spectrum.Thus,the structure of compound 5 was identified as 25,26,27-trinorcucurbita-5-ene-3β-ol-24-acid (Table 3).
Analysis of HRESIMS spectrum ascribed compound 6 to a molecular formula C26H44O2due to an adducting ion peak atm/z389.3442 [M+H]+.The NMR data (Table 2) of 6 exhibited one methylene less than those of 4,which can be confirmed by key1H-1H COSY correlations of H-21/H-20/H-22/H-23 as well as HMBC correlation from hydroxyl methyl proton atδH3.68 (2H,m) to C-20 (δC33.1).Thus,the structure of compound 6 was identified as a rare 24,25,26,27-tetranorcucurbitane triterpenoid,and named 24,25,26,27-tetranorcucurbita-5-ene-3β,23-diol.
The molecular formula of 7,C30H50O2,was determined due to HRESIMS adducting ion peak atm/z443.3904 [M+H]+.The1H NMR spectroscopic data (Table 2) gavetwo olefinic protons atδH5.59 and eight methyls atδH0.87 (d,6.5 Hz),0.72 (s),0.85 (s),0.78 (s),0.88 (s),0.89 (d,6.5 Hz),1.30 (s),1.31 (s).The13C NMR spectrum revealed the presence of 30 carbon resonances which were sorted into eight methyl carbons,nine methylenes,and seven methine carbons,and six quaternary carbons by DEPT NMR spectrum.Detailed comparison of the NMR data of 7 with those of maytefolin C [11] demonstrated that it possesses the same 18R-D:A-friedoeuphane skeleton,and differs from maytefolin C only at its side chain.The side chain of 7 was determined to be identical to that of known compound 9 by comparison of their1H and13C NMR chemical shifts (Table 2).This was further confirmed via the1H-1H COSY correlations of H-18/H-28,H-18/H-19/H-20 and the keyHMBC correlations from H-21,CH3-29,CH3-30 to C-22,and from H-20 to C-19 (Fig.2).The relative configurations of 7 were assigned as shown in Fig.3 by analysis of the NOESY spectrum (Fig.3).Thus,the structure of compound 7 was identified as 18R-D:A-friedoeuph-20-ene-22-ol-3-one.
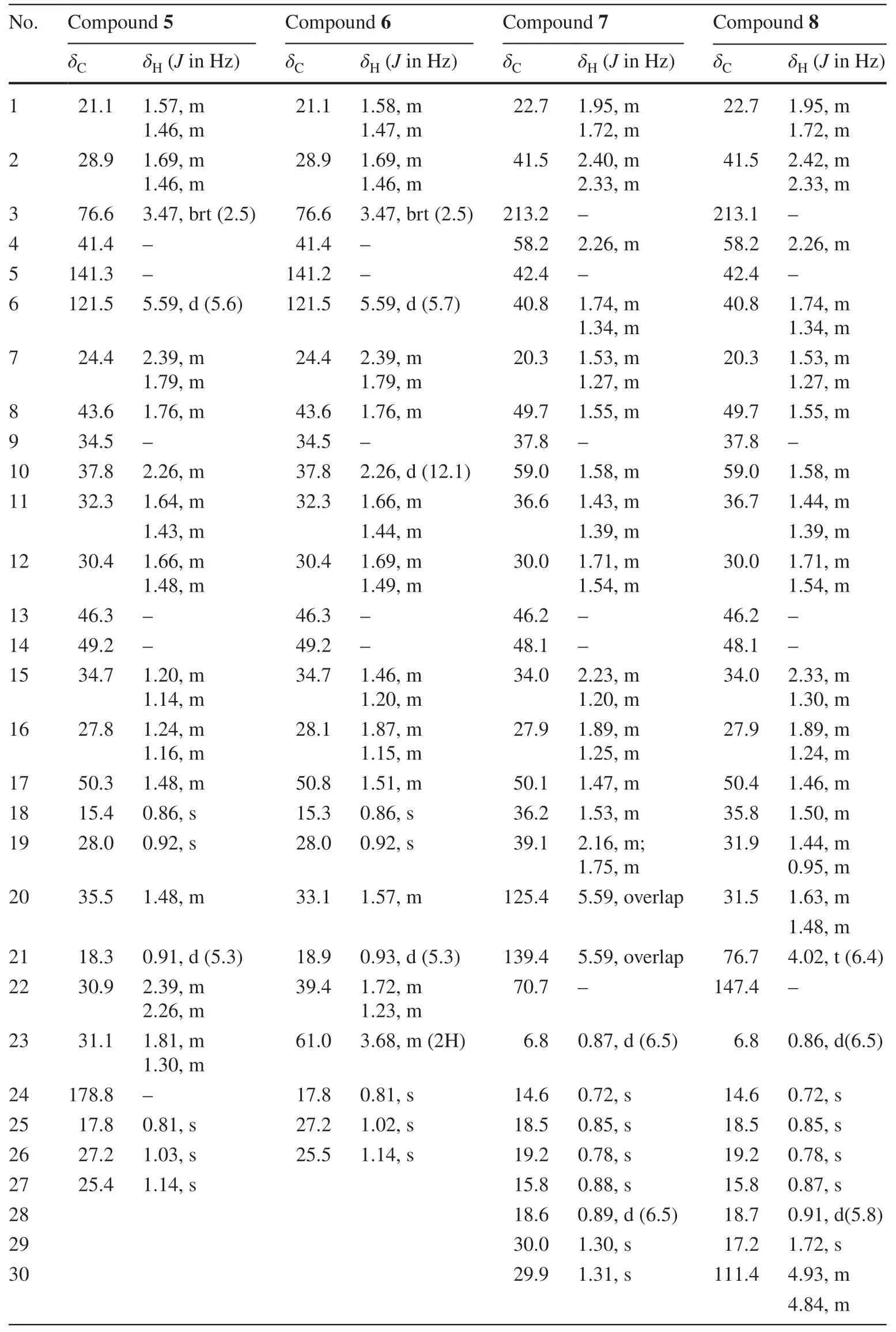
Table 2 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data of compounds 5-8 in CDCl 3
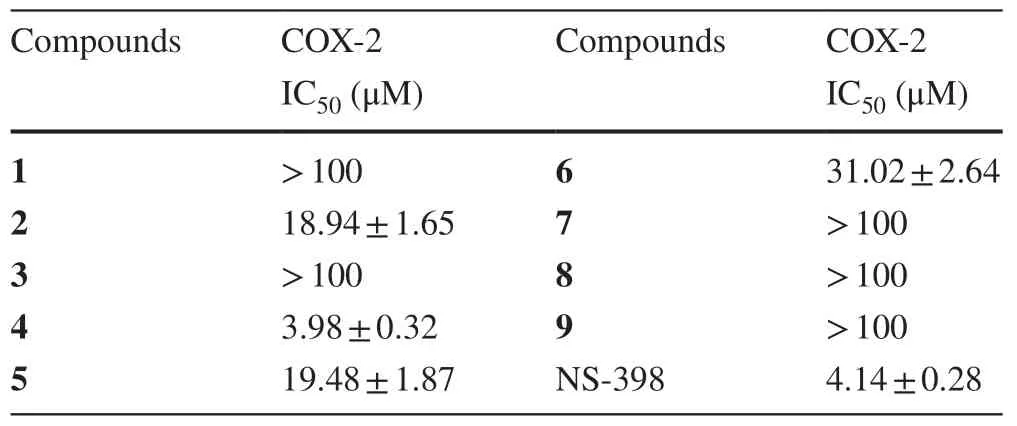
Table 3 Inhibitory effects of Compounds 1-9 against COX-2 in Vitro
Compound 8 was obtained as yellow solid,and had the same molecular formula as 7 (C30H50O2),as ascertained via HRESIMS adducting ion peak atm/z443.3924 [M+H]+.Detailed comparison of the NMR data with those of 7 revealed that 8 possessed a 18R-D:A-friedoeuphane skeleton as well,differing from 7 only in the positions of the double bond and the oxymethine at the side chain.The HMBC correlations from CH3-29 to two olefinic carbons atδC147.4 and 111.4 disclosed that a terminal double bond was placed at C-22 and C-30 positions.A hydroxyl was substituted at C-21 due to key HMBC correlations of CH3-29 and H-30 with the oxygenated methine carbon atδC76.7.The absolute configuration of C-21 was assigned asSon the basis of comparison of the chemical shifts of C-21 (δC76.7) and H-21 (δH4.02,1H,t,J= 6.4 Hz) with those in literature [12].Thus,the structure of compound 8 was identified as 18R-D:A-friedoeuph-22(30)-en-21S-ol-3-one.
All compounds were evaluated for their COX-2 inhibitory activities with NS-398 as a positive control.The results (Table 3) exhibited that compound 4 had the most potent inhibition against COX-2 with IC 50 values of 3.98 ± 0.32 μM,while compounds 2,5 and 6 showed mild inhibitory effects with IC50values of 18.94 ± 1.65,19.48 ± 1.87 and 31.02 ± 2.64 μM.Compounds 1-6 and 9 share similar or even the same rings A,B,C,D,and the major difference is their side chains.Therefore,it seems that the side chain is the main factor to influence the inhibitions of compounds 1-6 and 9 against COX-2.
3 Conclusion
In conclusion,this research led to the isolation of eight new triterpenoids and one known triterpenoid from theA.latifolia,in which compounds 4-6 are rare trinorcucurbitane or tetranorcucurbitane triterpenoids.It is the first report of cucurbitane-type triterpenoids from the genusAinsliaea.Interestingly,compound 4 showed potent inhibition against COX-2 with IC 50 values of 3.98 ± 0.32 μM.These results imply,except for sesquiterpenoids,triterpenoids may be another type of important chemical constituents being responsible for anti-inflammation inAinsliaeaspecies.Therefore,more attention should be paid to structural novel triterpenoids ofAinsliaeaplants.
4 Experimental Section
4.1 General Experimental Procedures
Optical rotations were measured on a PerkineElmer 341 polarimeter.1H and13C NMR spectra were recorded on Bruker Avance-500 spectrometers.ESI-MS were measured on an Agilent LC/MSD Trap XCT spectrometer,and HRESIMS were performed on an Agilent 6520 Accurate-MS Q-TOF LC/MS system.A preparative column (ZORBAX-ODS GSA10250AP1301,C18,5 μm,250 × 10 mm) was used for semi-preparative HPLC (Shimadzu LC-2010A HT).TLC analysis was run on HSGF254silica gel plates (10-40 μm,Yantai,China).Column chromatography (CC) was performed on silica (100-200,200-300 mesh,Yantai,China),YMC-GEL ODS-A (50 μm,YMC,Japan),Sephadex LH-20 (Amersham Pharmacia Biotech AB,Uppsala,Sweden).
4.2 Plant Material
The dried whole plants ofA.latifoliawere collected from Guiyang city of Guizhou province,PR China in September 2013,and authenticated by Prof.Long Qing-De,Department of Pharmacognosy,School of Pharmacy,Guiyang Medical University.An authentic specimen (No.20130905) was deposited at the School of Pharmacy,Second Military Medical University.
4.3 Extraction and Isolation
The dried whole plants ofA.latifolia(15.0 kg) were powdered and extracted with EtOH-H2O (80:20,v/v) twice at room temperature,48 h each time.The combined EtOH extracts were concentratedin vacuoto yield a crude extract (2.0 kg) which was then successfully partitioned with petroleum ether (PE),CHCl3,EtOAc,and MeOH,respectively,The CHCl3fraction (105 g) was chromatographed on a silica gel column,eluting with gradient PE/EtOAc (100:1;50:1;20:1;10:1;5:1) to give six fractions (F1: 19.2 g,F2: 5.2 g,F3: 7.3 g,F4: 21.7 g,F5: 7.9 g,F6: 13.1 g).Fraction F2 was subjected to column chromatography (CC) over Sephadex LH-20 (MeOH) and silica gel to give compounds 7 (12.0 mg),8 (4.2 mg).Fraction F3 was separated over Sephadex LH-20 (MeOH) followed by semi-preparative HPLC (CH3CN-H2O,100:0),to yield 1 (3.0 mg),2 (9.0 mg),and 3 (9.4 mg),respectively.Fraction F4 was subjected to ODS CC,eluted with a MeOH-H2O gradient,to yield 10 subfractions (F4A-F4 J).Subfraction F4B (507 mg) was subjected to CC over Sephadex LH-20 (MeOH) and silica gel to give compounds 4 (4.0 mg),5 (4.2 mg),6 (3.2 mg) and 9 (11.7 mg).
4.3.1 Cucurbita-5,22-diene-3β,24 S,25-triol (1)
White solid;[α]20D+18.7 (c0.10,CHCl3);UV (MeOH) λmax(log ε) 204 (3.71) nm;For1H NMR and13C NMR spectroscopic data,see Table 1;HRESIMSm/z493.3447 [M+Cl]-(calcd for C30H50O3,493.3454).
4.3.2 Cucurbita-5-ene-3β,24 S,25-triol (2)
White solid;[α]20D+46.6 (c0.30,CHCl3);UV (MeOH) λmax(log ε) 204 (3.72) nm;For1H NMR and13C NMR spectroscopic data,see Table 1;HRESIMSm/z495.3622 [M+Cl]-(calcd for C30H52O3,495.3610).
4.3.3 Cucurbita-5β,6β-epoxy-23-ene-3β,25-diol (3)
White solid;[α]20D+1.7 (c0.13,CHCl3);UV (MeOH) λmax(log ε) 201 (3.62),203 (3.69),231 (3.52) nm;For1H NMR and13C NMR spectroscopic data,see Table 1;HRESIMSm/z493.3457 [M+Cl]-(calcd for C30H50O3,493.3454).
4.3.4 Cucurbita-5-ene-3β,24-diol (4)
White solid;[α]20D+48.0 (c0.31,CHCl3);UV (MeOH) λmax(log ε) 205 (3.73),207 (3.71) nm;For1H NMR and13C NMR spectroscopic data,see Table 1;HRESIMSm/z403.3594 [M+H]+(calcd for C27H46O2,403.3571).
4.3.5 Cucurbita-5-ene-3β-ol-24-acid (5)
White solid;[α]20D+32.7 (c0.08,CHCl3);UV (MeOH) λmax(log ε) 203 (3.64) nm;For1H NMR and13C NMR spectroscopic data,see Table 2;HRESIMSm/z451.2980 [M+Cl]-(calcd for C27H44O3,451.2984).
4.3.6 Cucurbita-5-ene-3β,23-diol (6)
White solid;[α]20D+9.3 (c0.11,CHCl3);UV (MeOH) λmax(log ε) 205 (3.54) nm;For1H NMR and13C NMR spectroscopic data,see Table 2;HRESIMSm/z389.3442 [M+H]+(calcd for C26H44O2,389.3414).
4.3.7 18 R-D:A-friedoeuph-20-ene-22-ol-3-one (7)
White solid;[α]20D-17.4 (c0.37,CHCl3);UV (MeOH) λmax(log ε) 207 (3.18),231 (3.28) nm;For1H NMR and13C NMR spectroscopic data,see Table 2;HRESIMSm/z443.3904 [M+H]+(calcd for C30H50O2,443.3884).
4.3.8 18 R-D:A-friedoeuph-22-en-21 S-ol-3-one (8)
White solid;[α]20D-37.9 (c0.15,CHCl3);UV (MeOH) λmax(log ε) 201 (3.44),203 (3.54) nm;For1H NMR and13C NMR spectroscopic data,see Table 2;HRESIMSm/z443.3924 [M+H]+(calcd for C30H50O2,443.3884).
4.3.9 Cucurbita-5,23-diene-3 β,25-diol (9)
White solid,C30H50O2;1H NMR (500 MHz,CDCl3):δH0.79 (3H,CH3-30),0.85 (3H,s,CH3-18),0.87 (3H,d,J= 5.8 Hz,CH3-21),0.91 (3H,s,CH3-19),1.02 (3H,s,CH3-28),1.13 (3H,s,CH3-29),1.30 (2 × CH3,s,CH3-26,27),2.26 (1H,d,J= 12.1 Hz,H-10),2.38 (1H,m,H-7),3.47 (1H,br.t,J= 2.5 Hz,H-3),5.58 (3H,m,H-6,23,24);13C NMR (125 MHz,CDCl3):δC21.1 (t,C-1),28.9 (t,C-2),76.6 (d,C-3),41.4 (s,C-4),141.2 (s,C-5),121.4 (d,C-6),24.3 (t,C-7),43.6 (d,C-8),34.5 (s,C-9),37.8 (d,C-10),32.3 (t,C-11),30.3 (t,C-12),46.3 (s,C-13),49.2 (s,C-14),34.8 (t,C-15),27.8 (t,C-16),50.1 (d,C-17),15.4 (q,C-18),28.0 (q,C-19),36.2 (d,C-20),18.7 (q,C-21),39.1 (t,C-22),125.5 (d,C-23),139.4 (d,C-24),70.7 (s,C-25),29.8 (q,C-26),29.9 (q,C-27),17.8 (q,C-30),27.2 (q,C-28),25.4 (q,C-29);ESI-MS:m/z465 [M+Na]+(positive),441 [M-H]-(negative).
4.4 Determination of the Absolute Configuration of C-24 in Compounds 1 and 2
According to the published literature [9,10],a mixture of compound 1 (1.1 mg) and Mo 2 (OAc) 4 (1.2 mg) was prepared for CD measurement.The mixture was kept for 30 min to form a stable chiral metal complex,the CD spectrum of which was then recorded.The observed sign of the diagnostic ICD (induced CD spectrum) curve at around 315 nm was correlated with the absolute configuration of C-24 in compound 1.Compound 2 was also dealt with the same method as 1.
4.5 COX-2 Inhibitory Effect Assay
Cayman’s Colorimetric COX Inhibitor Screening Assay provides a convenient method for human recombinant COX-2 to screen isozyme-specific inhibitors.The assay measures the peroxidase component of COXs.The peroxidase activity is assayed colorimetrically by monitoring the appearance of oxidized N′,N,N,N′-tetramethyl-p-phenylenediamine (TMPD) at 590 nm.The COX-2 assay consisted of a 200 μL reaction mixture containing 150 μL assay buffer,10 μL Heme,10 μL COX-2,20 μL Colorimetric Substrate,and 10 μL test solution (1,5,10,20,80,100 μmol·L-1).The reactions were initiated by quickly adding 10 μL Arachidonic Acid,then incubating for 2 min at room temperature [13].
AcknowledgementsThe work was supported by NSFC (Nos.81573318,31870327,81230090,81520108030,1302658),National Major Project of China (No.2018ZX09731016-005),The Key Research and Development Program of China (Nos.2017YFC1702002,2017YFC1700200),Professor of Chang Jiang Scholars Program,Scientific Foundation of Shanghai China (Nos.17431902800,16401901300),Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (No.10DZ2251300).
Compliance with Ethical Standards
Conflict of interestThe authors declare that there are no conflicts of interest.
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creat iveco mmons.org/licen ses/by/4.0/),which permits unrestricted use,distribution,and reproduction in any medium,provided you give appropriate credit to the original author(s) and the source,provide a link to the Creative Commons license,and indicate if changes were made.
 Natural Products and Bioprospecting2020年1期
Natural Products and Bioprospecting2020年1期
- Natural Products and Bioprospecting的其它文章
- Dearomatized Isoprenylated Acylphloroglucinol Derivatives with Potential Antitumor Activities from Hypericum henryi
- UFLC-PDA-MS/MS Profiling of Seven Uncaria Species Integrated with Melatonin/5-Hydroxytryptamine Receptors Agonistic Assay
- Daphnane Diterpenoids from Trigonostemon lii and Inhibition Activities Against HIV-1
- Alkaloid Constituents of Ficus hispida and Their Antiinflammatory Activity
- A New Epi-neoverrucosane-type Diterpenoid from the Liverwort Pleurozia subinflata in Borneo