
Dearomatized Isoprenylated Acylphloroglucinol Derivatives with Potential Antitumor Activities from Hypericum henryi
2020-03-02 04:46YanSongYeManWuNaNaJiangYuanZhiLaoWenWeiFuXiaLiuXingWeiYangJuanZhang
Yan-Song Ye ·Man Wu ·Na-Na Jiang ·Yuan-Zhi Lao ·Wen-Wei Fu ·Xia Liu ·Xing-Wei Yang ·Juan Zhang ·
Hong-Xi Xu 2·Gang Xu 1
Abstract
KeywordsHypericum henryi ·Dearomatized isoprenylated acylphloroglucinols (DIAPs)·Apoptosis·Cell cycle arrest
1 Introduction
Natural phloroglucinol derivatives are widely distributed in Myrtaceae,Guttiferae,Euphorbiaceae,Aspidiaceae families as well as appeared in marine and microbial sources [1].In which prenylated acylphloroglucinols are a special kind of hybrid natural products originated from a polyketide combined isoprenylation biosynthetic pathways,and were mainly reported from the plants of genusHypericumandGarciniain the family Guttiferae [2-4].With their wide range of biological profiles and diverse molecular architectures exemplified by hyperforin [5],hypersubone B [6],hyperuralone A [7] and chinesins I/II [8],prenylated acylphloroglucinol derivatives have attracted great interest of chemists and pharmaceutists.
As a traditional folk medicine in China,Hypericum henryihas been used to treat hepatitis [9].Previous investigations on this plant have reported structurally diverse polycyclic polyprenylated acylphloroglucinols (PPAPs) such as hyphenrones A-X [10-12].As a part of systematic search on bioactive acylphloroglucinol derivatives,five new dearomatized isoprenylated acylphloroglucinols (DIAPs) derivatives,hyperhenols A-E (1-5) together with seven known analogues (6-12) were isolated fromH.henryi(Fig.1).In the bioactive study,compounds 1 and 6-8 were found to exhibit promising cytotoxic activities against three human cancer cell lines in vitro.And the further studies indicated compounds 6 and 7 could trigger autophagy,PINK1/Parkinmediated mitophagy in cancer cell lines,and also suppress lung cancer A549 cells metastasis targeting Akt and cofilin signaling pathways.In addition,6 and 7 also displayed significant anti-proliferation activities by inducing apoptosis and cell cycle arrest.Herein,the isolation,structure elucidation,and bioactivities evaluation of these compounds were reported.
2 Results andDiscussion
The MeOH extract was subjected to repeated column chromatography to yield five new DIAPs derivatives (1-5) together with seven known analogues hyphenrone J (6) [13],hyphenrone K (7) [13],hyperhenone E (8) [12],hyperhenone A (9) [12],hyperhenone B (10) [12],hyperhenone C (11) [12],and hyperhenone D (12) [12].
Hyperhenol A (1) was isolated as yellow oil and assigned molecular formula of C27H40O5with 8 degrees of unsaturation by HRESIMS (m/z443.2803 [M-H]-,calcd.C27H39O5,443.2803).The IR spectrum displayed bands for hydroxy (3417 cm-1) and carbonyl groups (1636 cm-1).The13C NMR data along with DEPT experiments showed 27 carbon signals including seven methyls,six methylenes,four methines,and ten quaternary carbons (three oxygenated tertiary carbons and two carbonyls).Detailed analysis of the13C NMR spectroscopic data (Table 1) indicated the presence of an isoprenyl (δC40.3,C-14;120.5,C-15;135.3,C-16;26.2,C-17;18.2,C-18),a DIAPs core including an enolized 1,3-diketone group (δC199.0,C-6;107.4,C-1;191.0,C-2),an enolic double bonds (δC111.1,C-3;179.4,C-4),a carbonyl (δC207.9,C-7),and a quaternary carbon atδC54.7 (C-5) [12,13].The location of the mentioned isoprenyl and the methyl (δC24.6,C-19) at C-5 was evidenced by the HMBC correlations from H 2-14 (δH2.62,2.56) and Me-19 (δH1.32) to C-4 (δC179.4),C-5 (δC54.7) and C-6 (δC199.0) (Fig.2).Besides the aforementioned DIAP moiety,the remaining 14 carbon signals can be attributed to asecbutyl group (δC43.4,C-8;δC17.1,C-9;δC27.7,C-10;δC12.4,C-11) and another C10unit.
Comparison of1H and13C NMR data indicated that the C10unit of 1 shared a similar structure with the monoterpenoid moiety of callistrilone B [14],which was confirmed by the1H-1H COSY correlations of H-3′/H-4′/H-5′/H-6′/H-1′,accompanied by HMBC correlations from Me-7′ (δH1.18) to C-1′ (δC42.7),C-2′ (δC73.3),and C-3′ (δC41.0);from Me-10′ (δH1.58) to C-8′ (δC151.0),C-5′ (δC47.1),and C-9′ (δC109.2);and from H2-9′ (δH4.71,4.67) to C-8′ (δC151.0),C-10′ (δC21.1),and C-5′ (δC47.1).The linkage of DIAP core and monoterpenoid moiety between C-1′ and C-3 was evidenced by HMBC correlations from H-1′ (δH3.24) toC-2 (δC191.0),C-3 (δC111.1),and C-4 (δC179.4) (Fig.2).Finally,thesec-butyl group only can be attached at C-1.
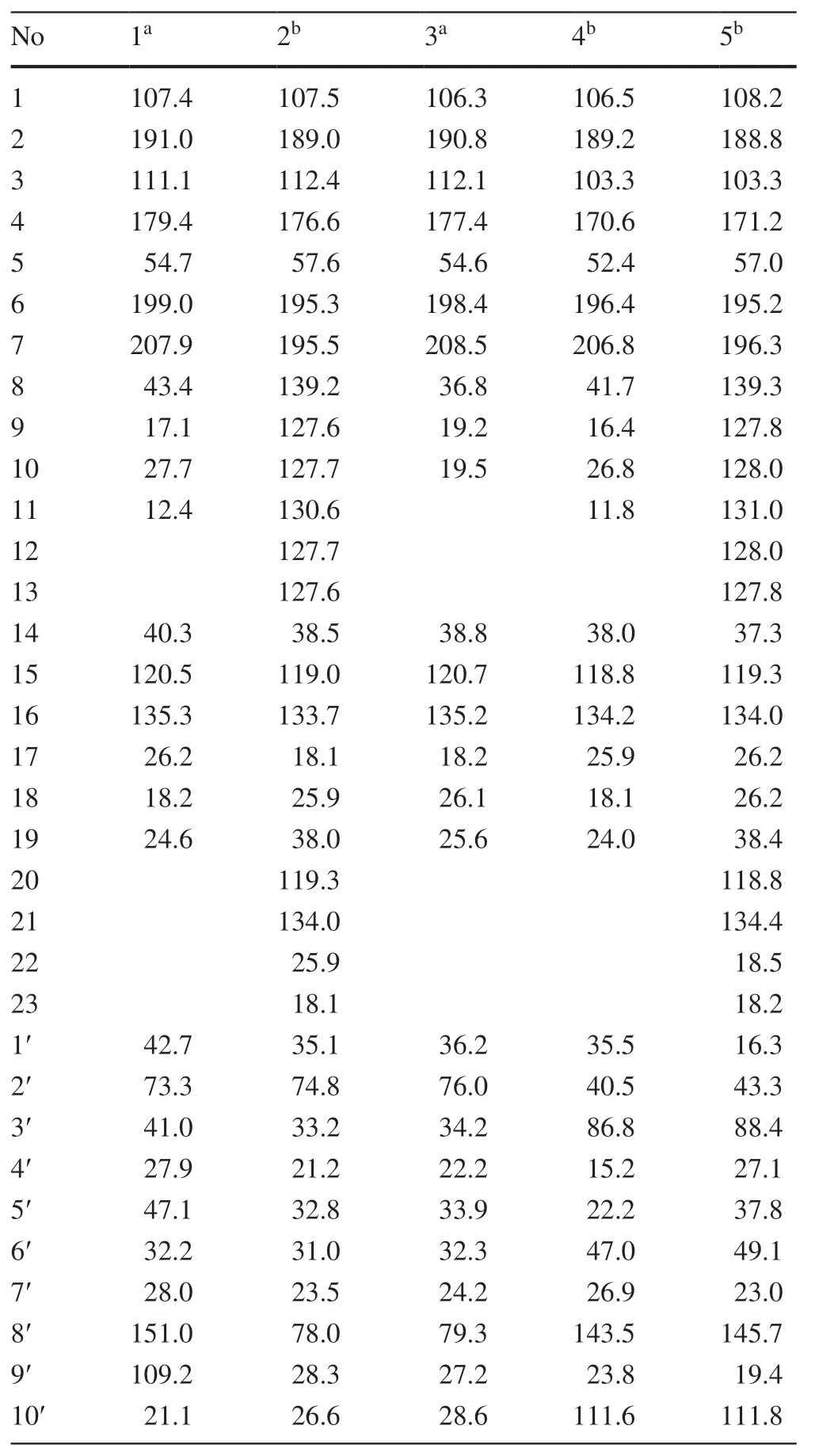
Table 1 The 13C (150 MHz) NMR data of compounds 1-5 (δin ppm)
The relative configuration of monoterpenoid moiety was determined by detailed interpretation of the ROESY spectrum.The NOE correlations of H-5′/H-1′ and H-1′/Me-7′ indicated that H-5′,H-1′ and Me-7′ were on the same side.However,due to the rotation of carbon-carbon single bond (C-3/C-1′) between DIAP core and monoterpenoid,determination of the configuration of C-5 is still challenging.For instance,an analogue of 1 (hyperhenone E,8),has also been reported with the configuration of C-5 undefined [12].In this study,hyperhenone E,as well as its crystals,was fortunately obtained,which unambiguously determined absolute configurations of 8 as 5S,1′R,2′R,5′S(Fig.3).Furthermore,the absolute configurations of C-5,C-1′,C-2′ and C-5′ in 1 were also determined to be the same with those of 8 via their well-matched ECD curves (Fig.4).
Hyperhenol B (2) was obtained as yellow oil.A molecular formula of C33H42O5,was deduced by its13C NMR and HRESIMS (m/z519.3106 [M+H]+,calcd.C33H43O5519.3105).The1H and13C NMR spectra of 2 and hyperhenone F are closely similar to each other [12].Comparative analyses of their NMR data revealed that the isopropyl in hyperhenone F was replaced by a phenyl,which was supported by the HMBC correlations from H-9/H-13 (δH7.43) to C-7 (δC195.5) and C-8 (δC139.2) in 2 (Fig.2).Because of the rigidity of the bicyclic skeleton,cyclohexane moiety tended to form boat conformation.Hence,the ROESY correlations of H-1′/H-3′ (δH1.96),H-4′/H-6′ (δH2.10),and H-6′/H-1′ showed the same orientations of H-1′ and Me-7′ (Fig.5).
Hyperhenol C (3) exhibited a molecular formula of C26H38O5,as assigned by HRESIMS (m/z429.2653 [M-H]-,calcd.C26H37O5,429.2646).The NMR spectra of 3 showed a close resemblance to those of hyperhenone F except that the signals for the isoprenyl at C-5 in hyperhenone F was replaced by a methyl in 3 [12],which can be further confirmed by the HMBC correlations from Me-19 (δH1.35) to C-5 (δC54.6),C-4 (δC177.4),C-6 (δC198.4) and C-14 (δC38.8).The similar NOE correlations of H-1′/H-3′ (δH1.70),H-3′/H-4′ (δH2.09),H-4′/H-6′ (δH1.61) and H-6′/H-1′ showed that relative configurations were the same as those of 2 (Fig.5).In the absence of sufficient evidence,configuration of C-5 could not be determined.
Hyperhenol D (4) was obtained as yellow oil.The molecular formula was established as C27H38O4based on its HRESIMS data (m/z427.2855 [M+H]+,calcd.C27H39O4427.2843),implying 9 indices of hydrogen deficiency.The characteristic information for a DIAPs core was clearly observed in the13C NMR spectra (δC106.5,C-1;δC189.2,C-2;δC103.3,C-3;δC170.6 C-4;δC52.4 C-5;δC196.4 C-6).A comparison of the 1D NMR data of 4 with those of chinesin I suggested that they shared closely similar plane structures [8].The molecular formulas (C27H40O4for chinesin I;C27H38O4for 4) revealed that 4 possessed one more degree of unsaturation [8],which could derived by the loss of H2O between hydroxyls of monoterpenoid and DIAPs core of chinesin I to afford 4.The ether linkage of C-4 and C-3′ was evidenced by indices of hydrogen deficiency,the downfield chemical shift of C-3′ (δC86.8) and the ROESY correlation of Me-7′/Me-17.The relative configurations of C-2′,C-3′,and C-6′ were elucidated by key ROESY correlations of Me-7′/H-2′,Me-7′/H-6′,and H-2′/H-6′.Unfortunately,the configuration of C-5 also could not be determined since the absence of sufficient evidence.
Hyperhenol E (5) was obtained as yellow oil,and its HRESIMS spectrum (m/z501.3008 [M+H]+,calcd.C33H41O4501.2999) showed a molecular formula of C33H40O4.The1H NMR data of 5 (Table 2) exhibited a monosubstituted benzene (δHH 7.30,3H;7.38,2H),two isoprenyl (δH4.75,t,J= 7.2 Hz;δH4.81,t,J= 7.2 Hz).The NMR spectra of 5 showed a close resemblance to those of 4 except for the replacements of thesec-butyl group at C-7 and the methyl at C-5 in 4 by a phenyl and an isoprenyl in 5,respectively.This conclusion was verified via the 1 H-1H COSY cross peak of H2-19/H-20 combined with the HMBC correlations of H2-19 (δH2.57 and 2.38) with C-4 (δC171.2)/C-5 (δC57.0)/C-6 and H-9 (δH7.30)/H-13 (δH7.30) with C-7 (δC195.2)/C-8 (δC108.2).In the ROESY spectrum,the obvious NOE correlation between Me-7′ and H-2′ can also be found as that in 4,but the diagnostic signals of H-2′/H-6′ and Me-7′/H-6′ in 4 were replaced by the H-2′/Me-9′and H-2′/H-10 (δH4.62),which indicated that the orientation of H-6′ was different with that of Me-7′/H-2′.
In the searching for their anticancer properties,compounds 1 and 6-8 were found to effectively inhibit cell growth in HeLa,A549,and MDAMB-231 cell lines (Table 3).Of which 6 and 7 could significant inhibit cancer cells growth with the IC50up to 0.07 and 0.09μM,respectively.Both the two compounds could also obviously increase mitochondrial fission and further activated the caspase-3,caspase-9,and increased PARP cleavage in HeLa cells (Fig.6 a,c).Treatment with 6 and 7 also increased the percentage of cells in G0/G1 phase and decreased in G2/M phase (Fig.6 b).Moreover,western blot results indicated that these two compounds efficiently suppressed the expression of cyclin D1 and Cdk 6 in HeLa cells,suggesting 6 and 7 induced cell cycle arrest.(Fig.6 c).Taken together,these results demonstrated that these compounds inhibited cell growth through inducing apoptosis and cell cycle arrest.
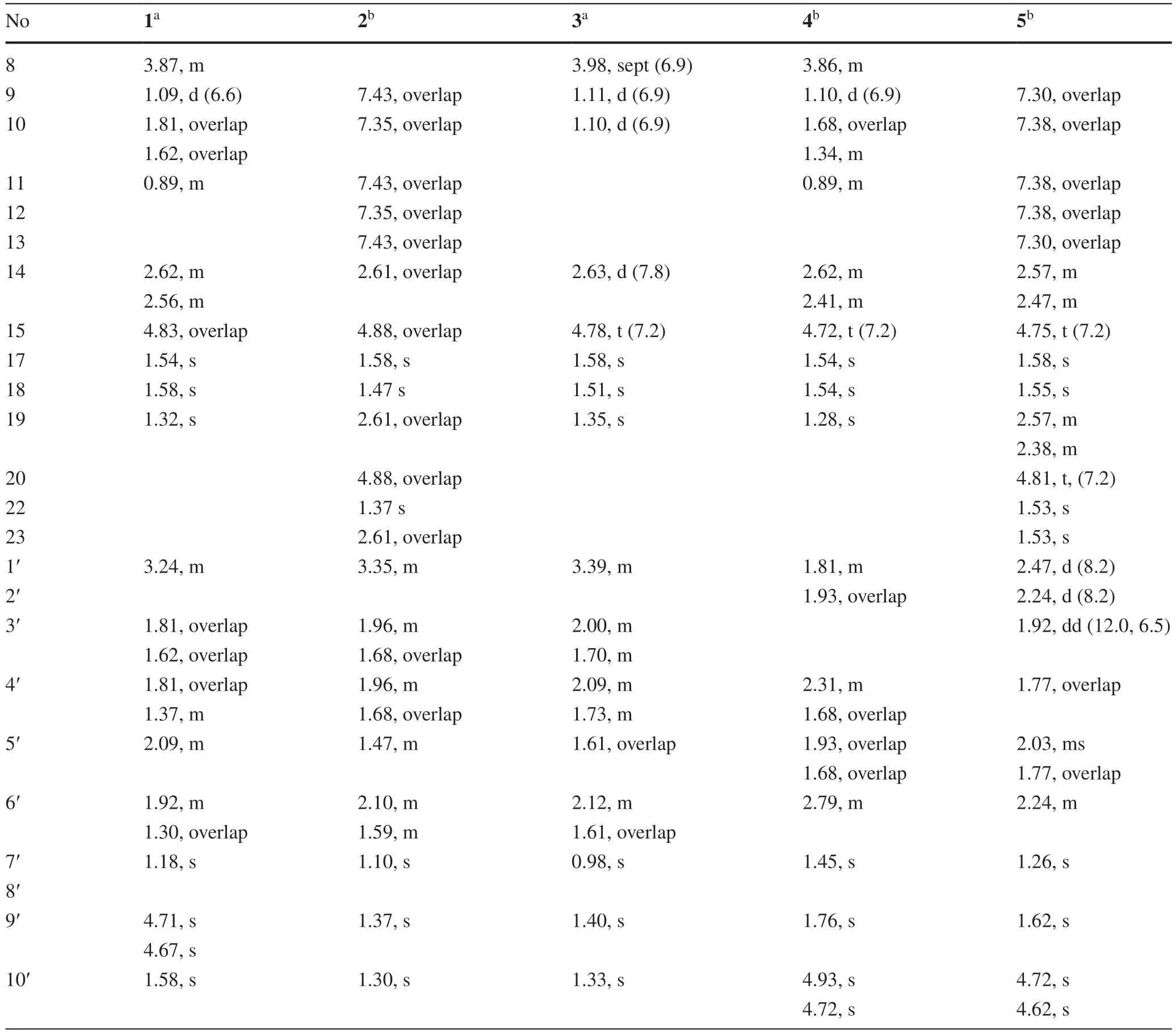
Table 2 The 1H (600 MHz) NMR data of compounds 1-5 (δ in ppm and J in Hz)
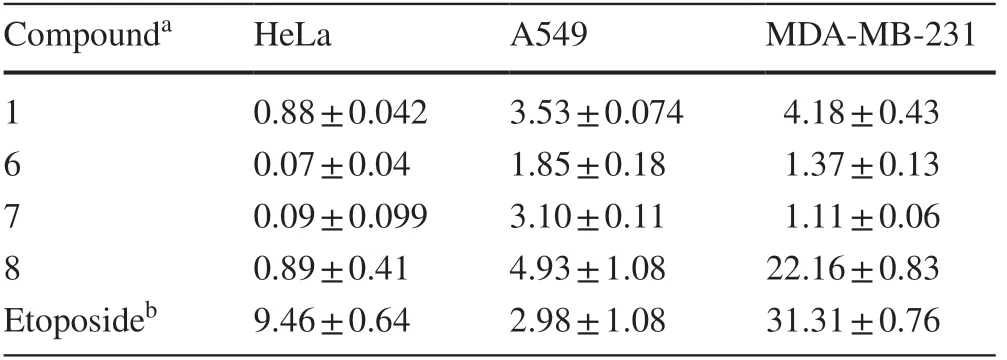
Table 3 Cytotoxicity of the isolates on three cancer cell lines with IC 50 values (μ M)
Autophagy is widely implicated in human diseases,offering a potential target for drug discovery [15].Then,the effects of 6 and 7 on autophagy were assessed.GFP-LC3 puncta were significantly increased upon these compounds treatment (Fig.7 a).Western blot analysis showed that 6 and 7 inhibited autophagy,as assessed by the increased expression of LC3 II and P62 (Fig.7 b).Similar to CCCP (mitophagy inducer) treatment,6 and 7 also increased the YFP-Parkin puncta formation (Fig.7 c).These data suggested that the compounds could induce PINK1/Parkinmediated mitophagy.In addition,the antimetastasis effects of these compounds were also studied.As shown in Fig.8,wound healing and migration assay suggested 6 and 7 could efficiently suppress cell metastasis consistent with sorafenib (SFB) treated,which also decreased the expression of vimentin,p-AKT and cofilin (Fig.8).Together,these results indicated that these isolates could suppress lung cancer A549 cells metastasis in vitro and may affect tumor metastasis targeted by Akt and cofilin signaling pathways.
In summary,five new and seven known DIAPs derivatives were isolated fromH.henryi.Structurally,these compounds were characterized by a dearomatized isoprenylated acylphloroglucinol core combined a functionalized cyclohexane or cyclopentane skeleton.It is worthy to note that several isolates exhibited significant cytotoxic activities in vitro.In addition,they also possess inducing autophagy,mitophagy,and anti-metastasis activities,which provided sufficient information on the potential application of these compounds on future drug development.Therefore,the finding of these DIAPs derivatives with potential antitumor properties may provide a new clue for the discovery of antitumor lead compounds,which should attract great interest from pharmacological and total synthetic communities.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 polarimeter.UV spectra were detected on a Shmadzu UV-2401PC spectrometer.IR spectra were determined on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr disks.All 1D and 2D NMR spectra were recorded on Bruker DRX-600 spectrometers using TMS as an internal standard.Unless otherwise specified,chemical shifts (δ) were expressed in ppm with reference to the solvent signals.ESIMS and HRESIMS analysis were carried out on Waters Xevo TQS and Aglient G6230 TOF mass spectrometers,respectively.Semi-preparative HPLC was performed on an Aglient 1100 HPLC with a ZORBAX SB-C18 (9.4 × 250 mm) column and a Waters 2695 HPLC with a CHIRALCEL OJ-RH column [4.6 × 150 mm cellulose tris-(4-methylbenzoate) coated on 5μM silica-gel].Silica gel (100-200,200-300 mesh,Qingdao Marine Chemical Co.,Ltd.,People’s Republic of China),and MCI gel (75-150μM,Mitsubishi Chemical Corporation,Tokyo,Japan) were used for column chromatography.Fractions were monitored by TLC (GF 254,Qingdao Marine Chemical Co.,Ltd.),and spots were visualized by heating silica gel plates sprayed with 10% H2SO4in EtOH.
3.2 PlantMaterial
The plants ofHypericum henryiwere collected in Dongchuan prefecture (Yunnan Province,People’s Republic of China) in September 2018.The plant was identified by ZHANG Yong-Zeng.A voucher specimen (No.2018H01) was deposited in Kunming Institute of Botany.
3.3 Extractionand Isolation
The sample (20.0 kg) was extracted with MeOH at room temperature and filtered,and the solvent was evaporated in vacuo.The crude extract was subjected to silica gel column chromatography eluted with CHCl3to afford a fraction (695.2 g).This fraction was separated over a MCI-gel column (MeOH-H2O from 7:3 to 10:0) to produce five fractions (Fr.A-E).Fr.A (262.3 g) was chromatographed on a silica gel column,eluted with petroleum ether-acetone (100:1 to 0:1),to yield five fractions (Fr.A1-A5).Fr.A2 (37.7 g) was separated over a RP-18 silica column (MeOH-H2O from 85:15 to 100:0) and obtained eleven fractions (Fr.A2-1-A2-11).Fr.A2-5 was purified by preparative TLC and semipreparative HPLC to afford 9 (12.3 mg),10 (11.5 mg) and 2 (10.8 mg).Fr.B (100 g) was chromatographed on a silica gel column,eluted with petroleum ether-ethyl acetate (50:1 to 0:1) to yield ten fractions (Fr.B1-B10).Fr.B3 (11.0 g) was purified by chromatograph on a silica gel column and preparative HPLC (MeOH-H2O,95:5) to afford 11 (25.9 mg) and 12 (4.7 mg).Fr.B4 (755.9 mg) and B6 (1.2 g) were further purified by prearative HPLC (MeOH-H2O,90:10) to afford 1 (15.1 mg),3 (13.3 mg),6 (26.5 mg) and 7 (12.0 mg).Fr.B2 (18.0 g) was separated over a RP-18 silica column (MeOH-H2O from 85:15 to 100:0),and obtained ten fractions (Fr.B2-1-B2-10) Compounds 4 (7.8 mg),5 (1.3 mg) and 8 (3.2 mg) were obtained from Fr.B2-2 by preparative HPLC and semipreparative HPLC.
3.3.1 Hyperhenol(1)
Yellow oil;[α]+250.8 (c0.35,MeOH);UV (MeOH)λmax(logε) 202 (4.14),225 (4.10),346 (4.02) nm;IR (KBr)νmax3417,2968,2932,1636,1520,1460,1337,1304,1233 cm-1;1H and13C NMR data,see Table s 1 and 2;ESIMSm/z443 [M-H]-;HRESIMSm/z443.2803 [M-H]-(calcd for C27H39O5,433.2803).
3.3.2 Hyperhenol(2)
Yellow oil;[α]+53.8 (c0.24,MeOH);UV (MeOH)λmax(logε) 203 (4.40),227 (4.20),343 (3.90) nm;IR (KBr)νmax3427,2969,2927,1623,1505,1448,1257,1202 cm-1;1H and13C NMR data,see Table s 1 and 2;ESIMSm/z519 [M+H]+;HRESIMSm/z[M+H]+;519.3106 (calcd for C33H43O5,519.3105).
3.3.3 Hyperhenol(3)
Yellow oil;[α]+199.0 (c0.31,MeOH);UV (MeOH)λmax(logε) 202 (4.08),228 (3.98),239 (3.97),281 (3.83),326 (3.91) nm;IR (KBr)νmax3431,2970,2932,2878,1651,1522,1470,1437 cm-1;1H and13C NMR data,see Table s 1 and 2;ESIMSm/z429 [M-H]-;HRESIMSm/z429.2653 [M-H]-;(calcd for C26H37O5,429.2646).
3.3.4 Hyperhenol(4)
Yellow oil;[α]+5.3 (c0.26,MeOH);UV (MeOH)λmax(logε) 320 (2.14),275 (2.30),241 (2.42),197 (2.50),310 (2.17),269 (2.30),215 (2.34) nm;IR (KBr)νmax3422,2970,2935,2876,1657,1618,1530,1462,1379 cm-1;1H and13C NMR data,see Table s 1 and 2;ESIMSm/z427 [M+H]+;HRESIMSm/z427.2855 [M+H]+(calcd for C27H38O4,426.2843).
3.3.5 Hyperhenol(5)
Yellow oil;[α]-45.0 (c0.12,MeOH);UV (MeOH)λmax(logε) 354(2.40),287(2.17),231(2.53),197(2.94),300(2.15),276(2.16),228(2.53),193(2.81) nm;IR (KBr)νmax3429,2967,2926,2854,1727,1659,1622,1587,1512,1448 cm-1;1H and13C NMR data,see Table s 1 and 2;ESIMSm/z501 [M+H]+;HRESIMSm/z501.3008 [M+H]+(calcd for C33H40O4,501.2999).
3.3.6 X-ray Crystallographic Analysis of Hyperhenone E(8)
C26H38O5,M= 430.56,a= 22.6496(4) Å,b= 9.4550(2) Å,c= 23.8898(4) Å,α= 90 °,β = 94.6410(10)°,γ = 90 °,V= 5099.27(16) Å3,T= 100(2) K,space groupP21,Z= 8,μ(CuKα) = 0.609 mm-1,56,500 reflections measured,17,671 independent reflections (Rint= 0.0269).The finalR 1values were 0.0363 (I> 2σ(I)).The finalwR(F2) values were 0.0966 (I> 2σ(I)).The finalR1values were 0.0366 (all data).The finalwR(F2) values were 0.0971 (all data).The goodness of fit onF2was 1.056.Flack parameter = 0.02(2).Crystallographic data for the structure of 8 have been deposited in the Cambridge Crystallographic Data Centre (deposition number: CDCC 1,941,889).
3.3.7 CellCulture
HeLa cells,GFP-LC3 HeLa cells,YFP-Parkin HeLa cells and A549 cells were maintained in DMEM (Gibco,D11527) supplemented with 10% fetal bovine serum,FBS (HyClone,SV30160.03) and 100 U/mL penicillin-streptomycin (Gibco/Invitrogen,15,140-122) in a humidified atmosphere containing 5% CO2at 37 °C.
3.3.7.1 MTT Assay and Determination of IC50The cells were seeded in a 96-well tissue culture plate at a predetermined density in 100μL of complete medium,attached overnight,and then treated with a series of concentrations of compound for 72 h.At the end of the incubation period,10μL MTT solution was added into each well of a 96-well plate for 4 h at 37℃.After the medium was removed,100μL DMSO was added to dissolve the purple crystals.After shaking for 5 min,the optical densities at 490 nm were measured using a Microplate Reader.
3.3.7.2 MitoTracker Red StainingHeLa cells were seeded on coverslips and treated with compounds 6 and 7 for 48 h.We then removed the media from the dish and added staining solution containing MitoTracker red (100 nM) incubation 30 min at 37 °C.The cells were fixed with 4% PFA in PBS for 15 min and observed using a fluorescence microscope.
3.3.7.3 Flow Cytometry AnalysisHeLa cells were treated with various concentrations of 7 and 8 for 48 h.Subsequently,the cells were harvested,washed with PBS and fixed with 70% alcohol at 4 °C overnight.Then cells were washed with PBS and stained with 20μg/mL PI/RNase staining buffer for 30 min and analyzed using FACSCalibur flow cytometer (Becton Dickinson,USA).
3.3.8 Immunofluorescence Microscopy
The GFP-LC3 or YFP-Parkin HeLa cells were treated with compounds for the indicated time point,and then the cells were fixed with 4% PFA in PBS for 15 min at room temperature.The cells were observed under a fluorescence microscope (Olympus,IX83).
3.3.9 Wound Healing Assay
Wound healing was used to evaluate cell motility as our previous study [16].Briefly,A549 cells were seeded into a 24-well culture plate.When the cells grew to 90% confluence,then a scratch was gently created through the cell monolayer by sterile 10μL pipette tips and loose cells were washed away.The cell migration was observed and imaged under an IX83 microscope for each condition and timepoint (0,48 h).(Olympus,Tokyo,Japan).
3.3.10 Cell Migration Assay
Cell migration assay were performed as described previously [17].In brief,cell migration was estimated using transwell chambers (Millicell,Germany) with a pore size of 8μM.For the migration assay,4.5 × 104 A549 cells resuspended in 100μL serum-free medium were seeded in the upper chamber with serum-containing medium in the lower chamber of 24-well transwell plates (BD Biosciences,San Jose,CA).After 24 h,the experiment was terminated by wiping the cells from the wells with a cotton swab and fixed and stained with 0.05% crystal violet for 20 min,scored under a light microscope in five random fields.
3.3.11 Western Blotting Analysis
Cells were harvested and lysed in a lysis buffer (62.5 mM Tris at pH 6.8,20% glycerol,2% SDS,phosphatase inhibitor),proteins were separated on SDS polyacrylamide gels and transferred to PVDF membranes (Millipore,Billerica,MA,USA).The membranes were blocked with 5% nonfat milk,and immunoblotted with primary antibodies at 4 °C overnight.After washed three times with TBST,membranes were incubated for 1 h with appropriate secondary antibodies at room temperature.The follow antibodies were used in our experiments: Caspase-3 (CST,9662),Cleaved-caspase-3 (CST,9661),Caspase-9 (CST,9502),PARP (CST,9542),LC3 (Sigma,L7543),P62 (BML,PM045),PINK1 (CST,6946),Tim23 (BD,611222),Tom20 (sc-17764),E-cadherin (CST,3195),Vimentin (CST,5741),pAKT (Ser473,CST,9171),AKT (CST,9272),Cofilin (CST,5175) and GAPDH (CST,5174).GAPDH was used as the loading control.Membranes were visualized with Image Quant LAS 4000 (General Electric Company).
AcknowledgementsThe work was financially supported by the NSFC-Joint Foundation of Yunnan Province (U1902213),Chongqing Municipal Natural Science Foundation (cstc2018jcyjAX0388),the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502),Southeast Asia Biodiversity Research Institute,CAS (2017CASSEABRIQG003),State Key Laboratory of Phytochemistry and Plant Resources in West China (P2017-KF02 and P2019-ZZ05),and the Natural Sciences Foundation of Yunnan Province (2019FA003).
Compliance with Ethical Standards
ConflictofinterestAll authors declare no conflict of interest.
OpenAccessThis article is licensed under a Creative Commons Attribution 4.0 International License,which permits use,sharing,adaptation,distribution and reproduction in any medium or format,as long as you give appropriate credit to the original author(s) and the source,provide a link to the Creative Commons licence,and indicate if changes were made.The images or other third party material in this article are included in the article’s Creative Commons licence,unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use,you will need to obtain permission directly from the copyright holder.To view a copy of this licence,visit http://creat iveco mmons.org/licen ses/by/4.0/.
 Natural Products and Bioprospecting2020年1期
Natural Products and Bioprospecting2020年1期
- Natural Products and Bioprospecting的其它文章
- Triterpenoidsfrom Ainsliaea latifolia and Their Cyclooxyenase-2(COX-2)Inhibitory Activities
- UFLC-PDA-MS/MS Profiling of Seven Uncaria Species Integrated with Melatonin/5-Hydroxytryptamine Receptors Agonistic Assay
- Daphnane Diterpenoids from Trigonostemon lii and Inhibition Activities Against HIV-1
- Alkaloid Constituents of Ficus hispida and Their Antiinflammatory Activity
- A New Epi-neoverrucosane-type Diterpenoid from the Liverwort Pleurozia subinflata in Borneo