
基于液相色谱-质谱技术的肾脏代谢组学分析方法研究
2020-02-28 13:31:12刘亚琪王中华何秉淑霍美玲傅文清再帕尔阿不力孜
分析测试学报 2020年1期
刘亚琪,王中华*,何秉淑,谢 冰,霍美玲,傅文清,周 帜,再帕尔·阿不力孜,2
(1.中央民族大学 生命与环境科学学院,生物成像与系统生物学研究中心,北京 100081;2.中央民族大学 药学院,北京 100081)
肾脏是人体重要的代谢和排泄器官,肾功能异常与多种疾病密切相关。目前,临床诊断肾脏疾病和药物肾毒性的常用指标为尿素氮(BUN)、血清肌酐(CR)和尿酸(Ua),但这些指标易受性别、年龄、体重等因素的影响[1],因此需要寻找特异性更强、灵敏度更高的生物标志物。代谢组学是研究生物体内源性小分子代谢物的整体及其变化规律的科学,是发现生物标志物的重要手段。目前,代谢组学在肾脏疾病诊断和药物肾毒性研究中已获得广泛应用。但已有研究多采用血液、尿液作为分析样本,肾脏组织代谢组学研究仍较少,而开展肾脏组织代谢组学研究有助于获得具有组织特异性的生物标志物。
肾脏组织中的代谢物具有种类繁多、结构多样、极性差异大等特点,对肾脏代谢组的全面分析提出了很大挑战。目前,液相色谱-串联质谱技术(LC-MS/MS)[2-6]因具有高灵敏度、高通量、强特异性的特点,已成为代谢组学研究中最常用的分析手段。代谢组学研究中提取组织样本的溶剂体系主要有甲醇/水、乙腈/水、甲醇/氯仿/水、甲醇/二氯甲烷/水[7-9]。但甲醇/水、乙腈/水只能提取强极性部分,获得的代谢物信息不够完整;甲醇/氯仿/水体系中,氯仿具有一定的毒性和环境污染性。另外,甲醇/二氯甲烷/水在提取时,强极性部分与弱极性部分上下层分离后,固体沉积物处于中间层,易造成对下层提取物的污染。此外,目前肾脏组织的代谢组学分析方法多采用单一的C18色谱柱进行色谱分离和分析,易造成强极性代谢物信息的缺失。因此,亟需建立一种对肾脏组织中小分子代谢物进行全面分析的代谢组学分析方法。
本研究以高灵敏、高分辨的液相色谱-质谱(LC-MS)技术为分析手段,采用甲基叔丁基醚/甲醇/水溶剂体系对大鼠肾脏组织中的代谢物进行提取,采用HILIC亲水色谱柱和反相C18色谱柱相结合的色谱分离系统对代谢物进行分离,并以10种代表性代谢物为研究对象,优化建立了适用于大鼠肾脏组织中小分子代谢物全面分析的代谢组学分析方法。该方法可同时获取肾脏组织中强极性和弱极性代谢物信息,具有灵敏度高、专属性强、稳定性好等特点,适用于肾脏疾病和药物肾毒性生物标志物的发现等研究。
1 实验部分
1.1 仪器与试剂
超高效液相色谱(Ultramate 3000)、Q-OT-qIT杂合型质谱仪(Orbitrap Fusion Lumos)、移液器(美国Thermo Fisher Scientific公司);冷冻干燥机与真空离心浓缩仪(德国Christ公司);离心机、混匀仪、涡旋仪(德国Eppendorf公司);高速分散机(德国IKA公司);0.22 μm滤膜(美国Agilent公司)。
牛磺酸(Taurine)、肌酸(Creatine)、L-异亮氨酸(L-Isoleucine)、1-甲基鸟嘌呤(1-Methylguanine)、5-甲硫腺苷(5-Methylthioadenosine);溶血磷脂酰胆碱:LysoPC(14∶0)、LysoPC(16∶0)、LysoPC(17∶0)、LysoPC(18∶0)、LysoPC(18∶2)标准品购于美国Sigma-Aldrich公司。乙腈、甲醇(德国Merck公司);甲酸(美国ROE公司);甲基叔丁基醚(MTBE,美国J.T.Baker公司);乙酸铵(美国Sigma-Aldrich公司);二氯甲烷(美国Mreda公司)。所有试剂均为色谱纯;实验用水为某品牌纯净水。
1.2 实验方法
1.2.1 样品采集与制备取SD大鼠肾脏,用生理盐水清洗表面血迹后立即置于液氮中冷冻保存,使用研钵在液氮中研磨成粉,粉末置于冷冻干燥机48 h。取冻干后的肾脏组织10 mg置于离心管中,加入400 μL预冻的冷溶剂(75%甲醇水),用高速分散机匀浆3 min(25 000 r/min,1 min;3 000 r/min,1 min;25 000 r/min,1 min)。超声10 min后加入1 mL甲基叔丁基醚,涡旋提取10 min(2 000 r/min)后,将样品置于4 ℃冰箱20 min,于4 ℃离心15 min(15 000 r/min),将上清液与蛋白和细胞沉淀物分离。上清液分为两层:甲基叔丁基醚/甲醇层(上层:弱极性代谢物)和甲醇/水层(下层:强极性代谢物),分别置于真空浓缩仪中浓缩至干,干燥时间分别约为2 h和1.5 h。取甲醇/水层干燥物用乙腈/水(80∶20,体积比)复溶至200 μL后离心、过滤,待分析;甲基叔丁基醚/甲醇层干燥物用乙腈/水(40∶60,体积比)复溶至120 μL后离心、过滤,待分析。
1.2.2 色谱条件强极性部分:采用Kinetex HILIC色谱柱(2.1 mm×150 mm,2.6 μm;Phenomenex公司);流动相为乙腈(A)和水(含5 mmol/L乙酸铵,B);流速为0.3 mL/min,进样量为10 μL,柱温为45 ℃。采用线性梯度洗脱:每次进样前用初始流动相平衡8 min,0~25 min,5%~40%B;25~30 min,40%~5%B。
弱极性部分:采用ACQUITY UPLC CSH C18色谱柱(2.1 mm×100 mm,1.7 μm;Waters公司);流动相为水(含0.1%甲酸,A)和乙腈(B);流速为0.25 mL/min,进样量为10 μL,柱温为40 ℃。采用线性梯度洗脱程序:每次进样前用初始流动相平衡5 min,0~2.5 min,2%~60%B;2.5~8 min,60%B;8~10 min,60%~81%B;10~18 min,81%~100%B;18~24.9 min,100%B;24.9~25 min,100%~2%B。
1.2.3 质谱条件强极性部分:离子源为电喷雾(ESI)源;采用正、负离子检测模式;扫描模式为全扫描;扫描范围:m/z100~1 000;质量分辨率:120 000;喷雾电压:3.8 kV(正离子模式),-3.0 kV(负离子模式);鞘气:0.20 MPa;辅助气:0.10 MPa;离子源温度:350 ℃(正离子模式),380 ℃(负离子模式);离子传输管温度:350 ℃。
弱极性部分:采用ESI源,正、负离子检测模式;扫描模式为全扫描;扫描范围:m/z100~1 000;质量分辨率:120 000;喷雾电压:3.8 kV(正离子模式),-3.0 kV(负离子模式);鞘气:0.40 MPa;辅助气:0.20 MPa;离子源温度:350 ℃(正离子模式),380 ℃(负离子模式);离子传输管温度:380 ℃。
1.2.4 数据处理获得正、负离子检测模式下的LC-(±)ESI-MS谱后,采用数据格式转换软件MSConvert将原始数据(.raw格式)转换成为mzXML格式文件,并导入数据处理软件XCMS进行峰识别、峰对齐、峰填充及峰过滤,最终获得包括质荷比(m/z)、保留时间(Retention time)及其峰面积的二维数据阵。将上述二维数据阵导入SIMCA-P进行主成分分析(PCA)。
2 结果与讨论
2.1 样品前处理方法的优化
代谢组学的样品前处理步骤对最终实验结果至关重要[10-11],提取代谢组信息时应尽可能兼顾代谢物不同的理化性质,最大程度地获得生物样本中包含的所有代谢物信息,保持代谢组的原始性和完整性,同时去除杂质,使待测物与分析技术相匹配,提高检测灵敏度。提取溶剂的种类、超声时间、提取时间等参数均会对实验结果产生影响,因此本研究对上述参数进行了优化。
2.1.1 提取溶剂的选择在文献调查的基础上[12-17],本研究以色谱峰数目、代表性代谢物的峰面积以及提取平行性为指标,比较了二氯甲烷/甲醇/水(28∶41∶42,体积比)、MTBE/二氯甲烷/甲醇/水(63∶36∶30∶35,体积比)、MTBE/甲醇/水(100∶30∶35,体积比)作为提取溶剂时对肾脏组织中代谢物的提取效果。
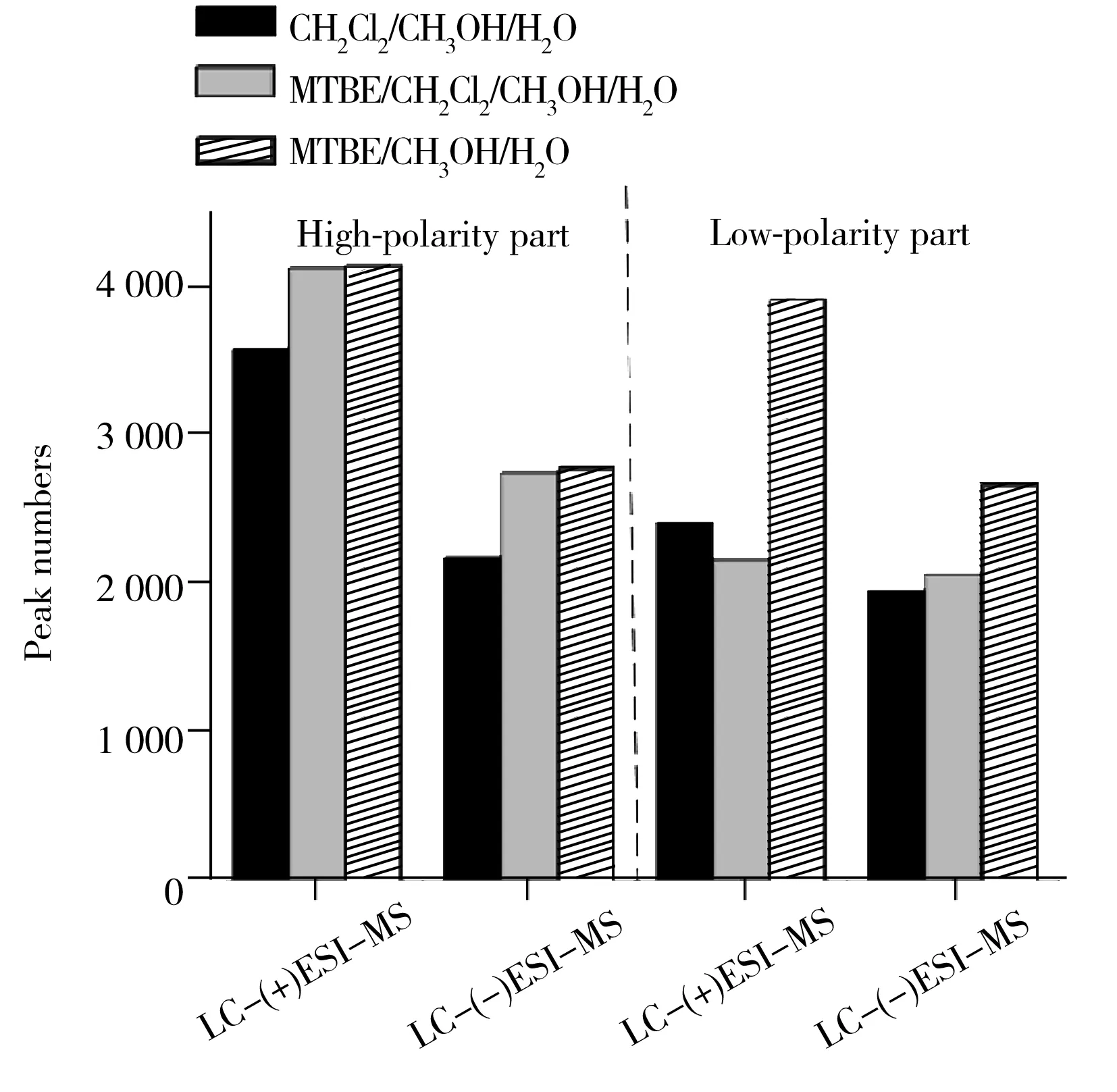
图1 不同溶剂提取获得的色谱峰数目比较
首先,将正、负离子检测模式下获得的LC-MS原始谱图数据经格式转化及XCMS软件进行预处理,得到不同溶剂体系提取检测到的色谱峰数目(如图1)。结果显示,MTBE/甲醇/水溶剂体系提取得到的色谱峰数目最多(共得到13 556个色谱峰,其中强极性部分6 956个,弱极性部分6 600个),其次是MTBE/二氯甲烷/甲醇/水溶剂体系(共得到11 125个色谱峰,其中强极性部分6 890个,弱极性部分4 235个),二氯甲烷/甲醇/水溶剂体系提取得到的色谱峰数目最少(共得到10 152个色谱峰,其中强极性部分5 773个,弱极性部分4 379个)。
随后对不同提取溶剂体系获得的10种代表性代谢物的提取离子流色谱峰面积进行了比较,结果如图2所示。由图2可知,MTBE/甲醇/水溶剂体系对肾脏中LysoPC(14∶0)、LysoPC(16∶0)、LysoPC(17∶0)、LysoPC(18∶2)和LysoPC(18∶0)等弱极性代谢物的提取效果显著优于其他两种溶剂体系,对L-异亮氨酸、牛磺酸、肌酸、1-甲基鸟嘌呤和5-甲硫腺苷等强极性代谢物的提取效果也最优,但与其他两种溶剂体系的差别较小。
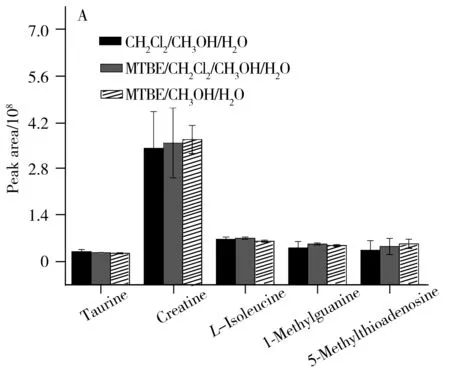
进一步计算了各代表性代谢物提取离子流色谱峰面积的相对标准偏差(RSD),考察了不同提取溶剂体系的提取平行性,结果见表1。从表中可以看出,MTBE/甲醇/水溶剂体系提取得到的各代谢物色谱峰面积的RSD最小,大部分代谢物的RSD值小于15%,表明该溶剂体系对肾脏中代谢物的提取平行性最好。
综上所述,本研究最终选择MTBE/甲醇/水(100∶30∶35)作为提取溶剂。

表1 不同溶剂提取物LC-(+)ESI-MS分析获得的代表性代谢物色谱峰面积的相对标准偏差
2.1.2 组织匀浆对提取效果的影响通过比较不匀浆与匀浆3 min条件下所获得的色谱峰数目,考察了组织匀浆对于代谢物提取效率的影响。结果显示,组织匀浆条件下,强极性部分和弱极性部分检测到的色谱峰数目均显著高于不匀浆条件下获得的结果。上述结果表明,组织匀浆可显著提高代谢物的提取效率。因此本实验采用高速分散机对肾脏组织粉末匀浆后再进行溶剂提取,并选择在冰浴条件下进行组织匀浆以防止提取液温度过高对代谢物造成破坏,具体程序如“1.2.1”所示。
2.1.3 超声时间的选择以提取到的色谱峰数目为指标,考察了不同超声时间(0、5、10、20 min)对代谢物提取效果的影响。结果表明,超声0、5、10、20 min时,强极性部分可分别提取5 976、6 419、6 313和6 376个色谱峰,弱极性部分可分别提取5 391、5 619、6 132和6 272个色谱峰,共可分别提取11 367、12 038、12 445和12 648个色谱峰,表明超声20 min提取的色谱峰数目最多,但与10 min差别不明显。为避免超声时间过长产生热量而导致代谢物信息发生改变,最终选择超声提取时间为10 min。
2.1.4 涡旋提取时间的选择本研究在代谢物提取过程中采用涡旋进行辅助提取,并比较了不同涡旋时间(5、10、20 min)获得的色谱峰数目。结果表明,涡旋提取5、10和20 min时,强极性部分可分别提取5 594、6 257和6 357个色谱峰,弱极性部分可分别提取5 349、5 541和6 339个色谱峰,共可分别提取10 943、11 798和12 696个色谱峰。综合考虑,最终选择涡旋时间为20 min。
2.2 色谱与质谱条件的优化
2.2.1 色谱条件的优化强极性部分选用Kinetex HILIC色谱柱(2.1 mm×150 mm,2.6 μm),弱极性部分选用ACQUITY UPLC CSH C18色谱柱(2.1 mm×100 mm,1.7 μm),分别考察了进样体积、流速、梯度洗脱程序、柱温等对色谱分离效果的影响。优化后的总离子流色谱图(TIC)如图3所示,强极性部分在进样量为10 μL,流速为0.30 mL/min,梯度洗脱时间为30 min,柱温为45 ℃的条件下,各色谱峰的分离效果良好;弱极性部分在进样量为10 μL,流速为0.25 mL/min,梯度洗脱时间为25 min,柱温为40 ℃的条件下,各色谱峰的分离效果良好。
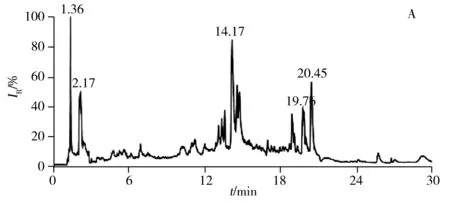
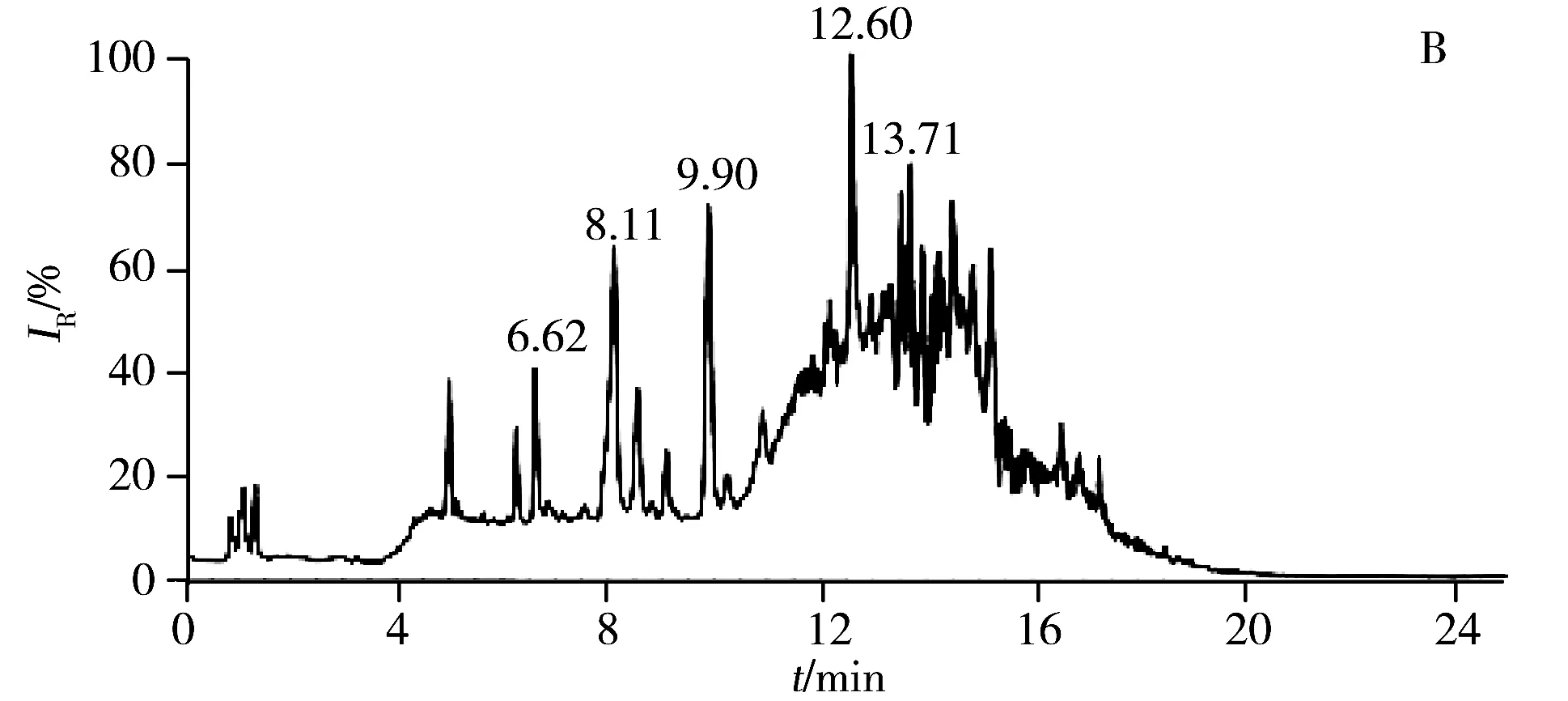
图3 LC-(+) ESI-MS分析获得肾脏代谢物的总离子流色谱图
2.2.2 质谱条件的优化样品测试前,采用质谱校正液将质谱的质量精度校正在2个ppm以内。选择牛磺酸、肌酸、L-异亮氨酸、1-甲基鸟嘌呤、5-甲硫腺苷、LysoPC(14∶0)、LysoPC(16∶0)、 LysoPC(17∶0)、LysoPC(18∶0)和LysoPC(18∶2)10种对照品的混合溶液,采用蠕动泵直接进样分析,对离子源喷雾电压、鞘气、辅助气体、离子源温度、离子传输管温度等质谱参数进行了优化,以获得最大的质谱响应和检测灵敏度。
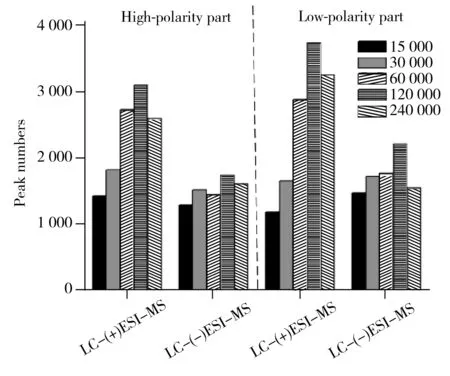
图4 肾脏提取物在不同质量分辨率条件下经LC-(±)ESI-MS检测到的色谱峰数目
此外,本研究发现质谱的质量分辨率对方法的灵敏度和特异性有显著影响[18]。肾脏提取物在不同质量分辨率条件下(15 000、30 000、60 000、120 000和240 000)进行LC-(±) ESI-MS分析检测到的色谱峰数目如图4所示。结果显示,当质量分辨率从15 000增至120 000时,测得的色谱峰数目逐渐增多。推测可能是由于随着质量分辨率的提高,色谱未完全分离的分子量相近的共流出物在质谱中获得了分离。图5为肾脏提取物弱极性部分在LC-(+) ESI-MS检测模式下保留时间为8.26 min时共流出物的质谱图,由图可知,质量数相差0.03 Da的共流出物在质量分辨率为15 000的条件下无法实现质谱分离,随着质量分辨率的提高,其分离情况显著改善,从而可提高其检测的特异性、信噪比和灵敏度。但当分辨率从120 000增至240 000时,测得的色谱峰数目有所降低,推测可能与Orbitrap型高分辨质谱仪的扫描速度会随着质量分辨率的增加而降低有关。综合考虑特异性和灵敏度,选择质谱分辨率为120 000。最终优化的质谱条件如“1.2.3”所示。
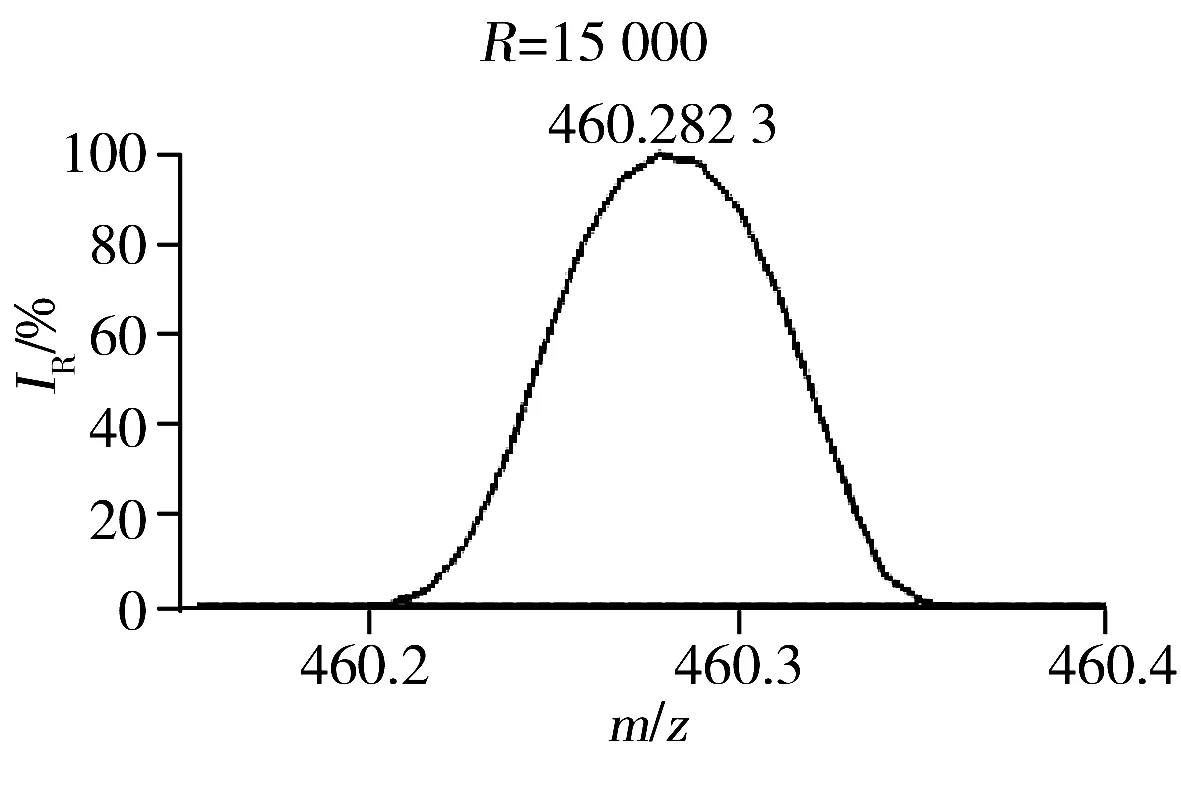
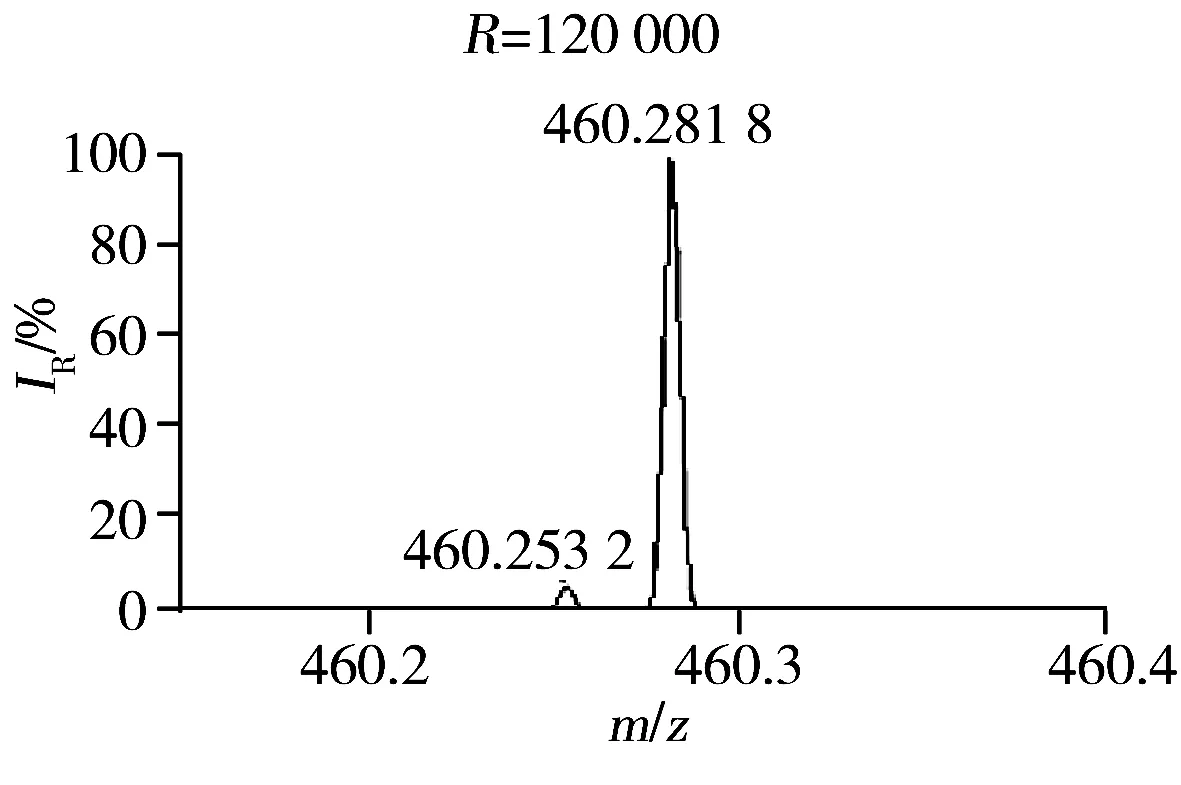

图5 肾脏提取物弱极性部分在LC-(+) ESI-MS检测模式下保留时间为8.26 min时共流出物的质谱图
2.3 方法学评价
2.3.1 仪器精密度取大鼠肾脏的冻干粉末,按“1.2.1”方法进行提取,对同一样品连续进样6次进行分析,分别计算10种代表性代谢物色谱保留时间和提取离子流色谱峰面积的RSD,对仪器精密度进行考察。结果显示,各代谢物提取离子流色谱峰面积的RSD均小于10%(表2)。此外,各代谢物保留时间的RSD值小于5.0%。上述结果表明仪器的精密度良好。
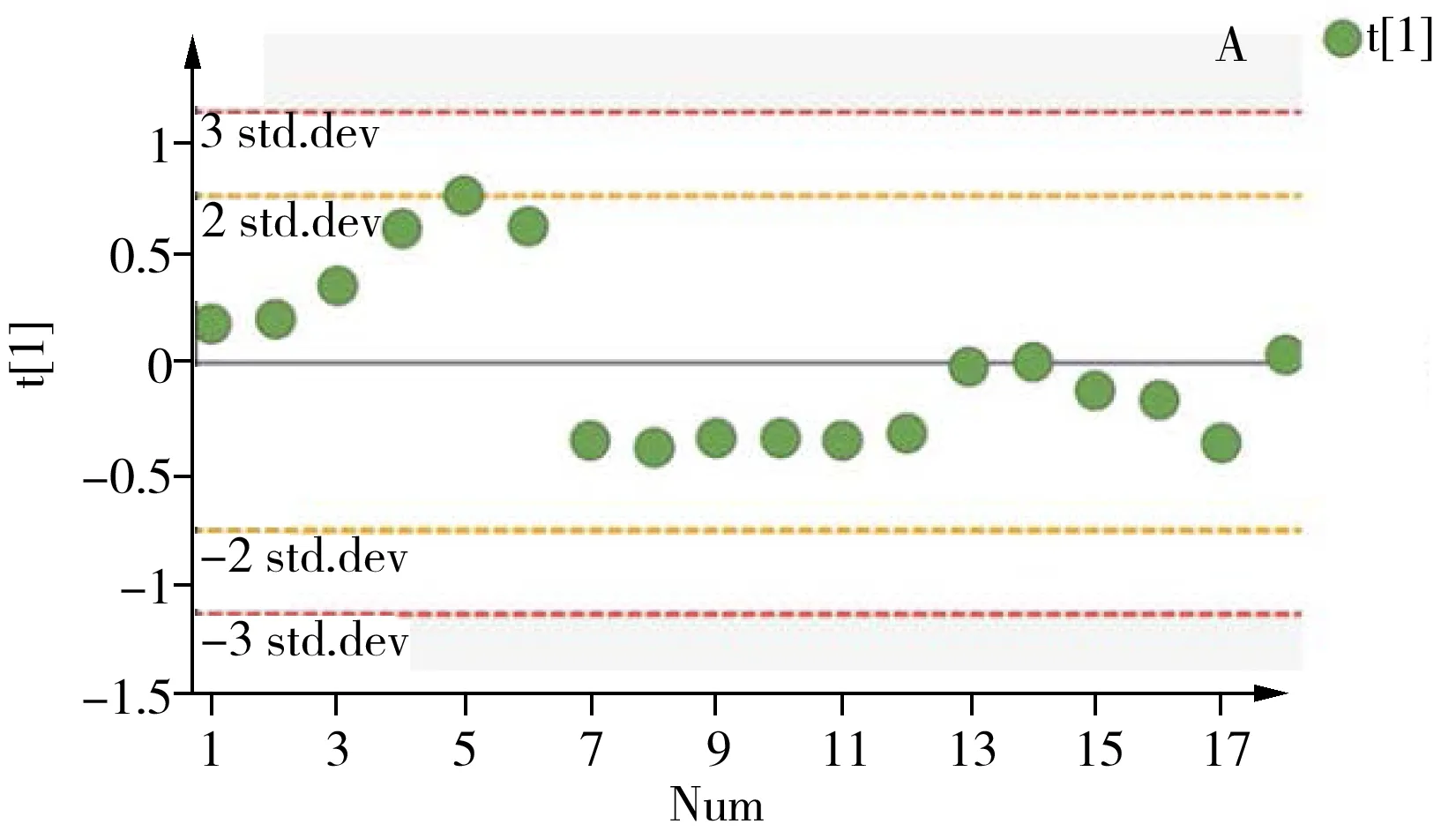
2.3.2 方法精密度取大鼠肾脏的冻干粉末,按“1.2.1”方法进行提取,平行制备QC样本,每天进行6样本分析,连续测定3天,将获得的数据进行PCA分析,同时计算不同极性的内源性代谢物提取离子流色谱峰面积的RSD值,以评估方法的批内和批间精密度。经PCA分析后各样本在第一主成分上的投影结果如图6所示,18个QC样本的偏差均在2SD范围内。各代谢物提取离子流色谱峰面积的批内和批间RSD均不大于15%(表2),此外保留时间的批内和批间RSD小于15%。上述结果表明,所建立的方法具有良好的重复性,从而可保证测试样本间的差异主要来源于不同处理组样品中内源性代谢物的差异,而不是由分析方法的误差产生,因此本方法可用于肾脏代谢组学研究。
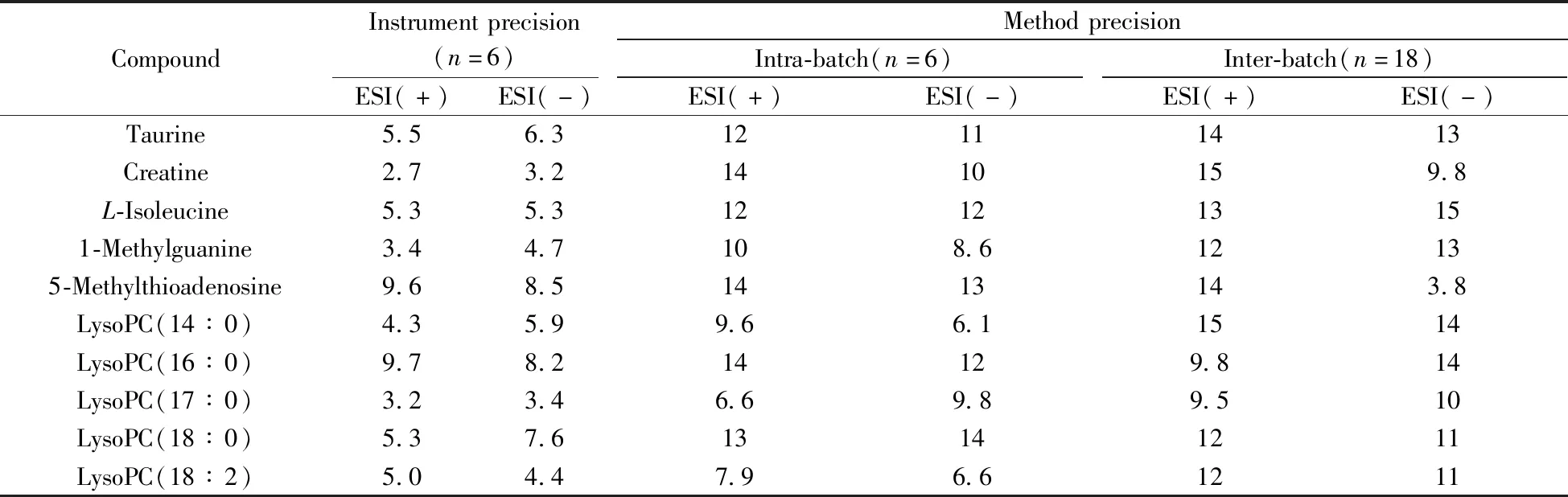
表2 LC-(±) ESI-MS方法的仪器精密度和方法精密度
3 结 论
本研究建立了基于液相色谱-超高分辨质谱技术的肾脏代谢组学分析方法。该方法以MTBE/甲醇/水溶剂体系作为提取溶剂,采用亲水色谱和反相色谱相结合的色谱分离系统对代谢物进行分离,可同时获取肾脏组织中的强极性和弱极性代谢物信息。方法学考察结果表明,该方法具有灵敏度高、专属性强和稳定性好等特点,可为肾脏疾病和药物肾毒性生物标志物的发现提供一种新方法。目前,该方法已应用于药物肾毒性代谢组学研究中。
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:36:10
时代英语·高一(2019年5期)2019-09-03 02:09:34
植物研究(2018年4期)2018-07-24 00:52:26
癌变·畸变·突变(2016年5期)2016-08-22 05:55:20
电测与仪表(2016年11期)2016-04-11 12:20:42
分析测试学报(2015年7期)2016-01-13 06:19:16
电源技术(2015年5期)2015-08-22 11:18:28
质谱学报(2015年5期)2015-03-01 03:18:37
中医研究(2014年11期)2014-03-11 20:29:54
湖南造纸(2013年1期)2013-07-10 01:06:40
