
采用偏最小二乘法的基因-药物共模块识别
2020-02-27 09:41毛玉杰魏东李玉双
华侨大学学报(自然科学版) 2020年1期
毛玉杰, 魏东,2, 李玉双
(1. 燕山大学 理学院, 河北 秦皇岛 066004;2. 河北数港科技有限公司, 河北 秦皇岛 066004)
近年来,国际上开展了一系列大型的药物筛选实验,其中,规模最大的两个研究项目是癌症细胞系百科全书(CCLE)[1]和癌症基因组项目(CGP)[2],它们分别识别与单个药物有关联的基因组分子特征.然而,在癌症的治疗中,往往都是多个药物联合使用,因为一个药物对应的靶标基因不止一个,同时,一个靶标基因所靶向的药物也不止一个[3].通过探索多个药物与多个基因之间的联系,即模块化分析,可以更直观地发现高维数据之间隐含的生物信息.胡尊胜等[4]基于复杂网络,研究蛋白质界面网络中的模体和模块,有利于研究蛋白质界面网络的形成机制;Zhang等[5]提出稀疏网络正则约束的非负矩阵分解模型,对microRNA-基因共模块进行识别;Chen等[6]提出带有网络正则约束的稀疏偏最小二乘模型(SNPLS),对基因-药物共模块进行识别,通过对各模块的分析,有利于发现不同药物之间相互作用的分子机理.受SNPLS模型的启发,本文提出伴有基因和药物关联网络正则约束的稀疏偏最小二乘(SGDPLS)算法,并将其应用于癌症药物敏感性基因组学数据库中.
1 材料和方法
1.1 数据来源和预处理
从癌症药物敏感性基因组学数据库(genomics of drug sensitivity in cancer,GDSC)中下载了细胞系的基因表达数据X1∈R641×13 321和细胞系药物响应数据X2∈R641×95.从Pathway Commons数据库(http:∥www.pathwaycommons.org/)中下载基因关联网络A,该网络对应的邻接矩阵为A.从NCBI PubChem数据库中下载药物的化学结构SDF文件,使用OpenBabelGUI工具箱读取药物SDF文件,并将药物特征转化为分子指纹,通过计算Jaccard相关系数[7],得到药物相似性矩阵D.将D中小于0.8的位置赋值为零,其余位置赋值为1,得到药物关联网络B,该网络对应的邻接矩阵为B.为了保证数据都处于同一数量级,且避免离群数据的出现,对模型中的初始数据X1和X2作了Z-score处理.
1.2 伴有基因和药物关联网络正则约束的稀疏偏最小二乘算法
在SNPLS算法的基础上,增加了药物关联网络信息,构建SGDPLS算法,其流程图如图1所示.图1中:H是药物Docetaxel对应的化学结构;目标函数(1)中的L和C是由邻接矩阵A和B经过拉普拉斯矩阵变换得到的[8].SGDPLS算法对权向量g和d进行稀疏约束,可以使模型在迭代优化过程中不陷入过拟合状态,增加识别模块的可解释性.
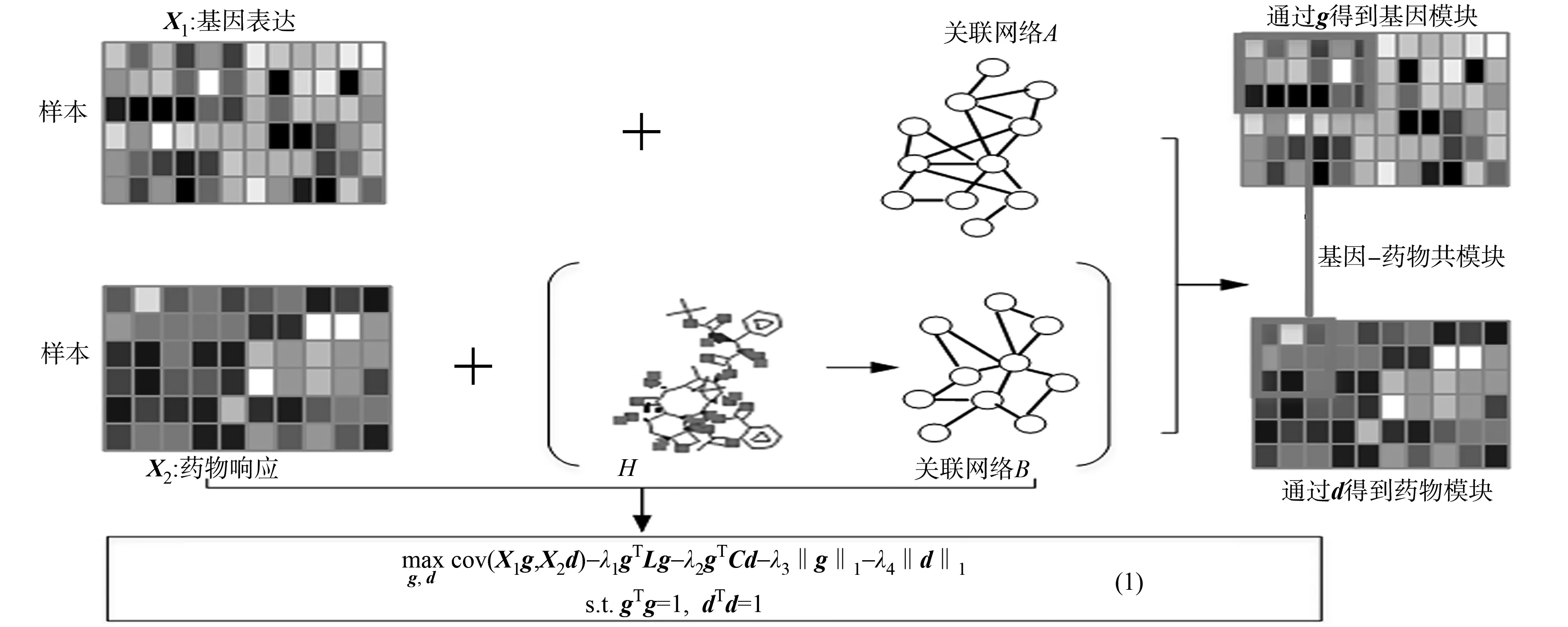
图1 SGDPLS算法流程图Fig.1 SGDPLS algorithm flow chart
1.3 SGDPLS模型的求解
图1中的目标函数(1)是一个非凸函数,利用传统的优化算法很难得到模型的全局最优解.Chen等[6]采用交替坐标下降法,通过交替更新g和d,求得模型的局部极大值.文中将该方法用于SGDPLS模型的求解,SGDPLS模型的求解过程有以下3个步骤.

步骤2交替更新g和d.
固定变量g,更新变量d,则
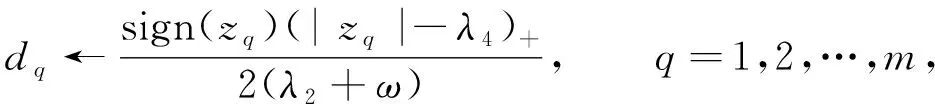
v=X2d.
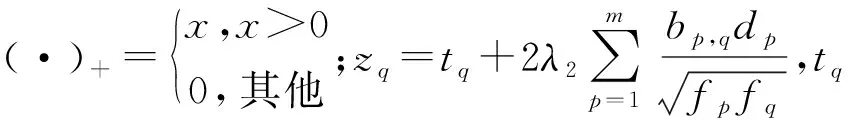
固定变量d,更新变量g,则
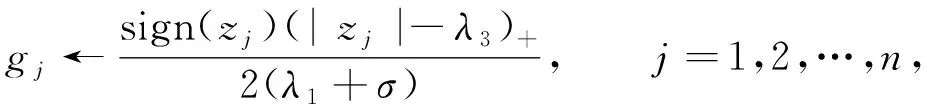
u=X1g.
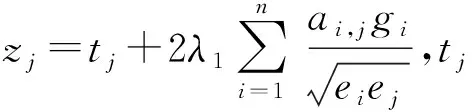
步骤3重复步骤2直到u收敛,算法终止.
对目标函数(1)做上述迭代运算,得到权向量g和d识别共模块之前,先对g和d作Z-score标准化处理.通过设定阈值T,筛选共模块的成员.对于基因(药物)模块,选取g(d)中比阈值T1(T2)大的位置所对应的基因(药物)作为基因(药物)模块的成员.对于样本模块,首先计算u=X1g和v=X2d;然后,对u+v作标准化处理,取向量中比阈值T3大的位置所对应的样本作为样本模块的成员.在筛选样本、基因和药物共模块时,设置的阈值分别为T1=3,T2=3,T3=2.阈值过大(小),会使识别出的共模块过小(大).模块过大,会导致模块中包含的信息太多,有用的信息不易被挖掘出来;模块过小,则包含的信息过少,不具有生物可解释性[4].
2 结果分析
将SGDPLS模型用于GDSC数据库,通过MATLAB编程[10]调参,识别出20个不同的基因-药物共模块.SGDPLS模型识别共模块分布图,如图2所示.由图2可知:有17个样本模块其样本个数分布在24~34范围内(图2(a));有13个基因模块其基因个数分布在111~151范围内(图2(b));12个药物模块的药物个数为2(图2(c)).
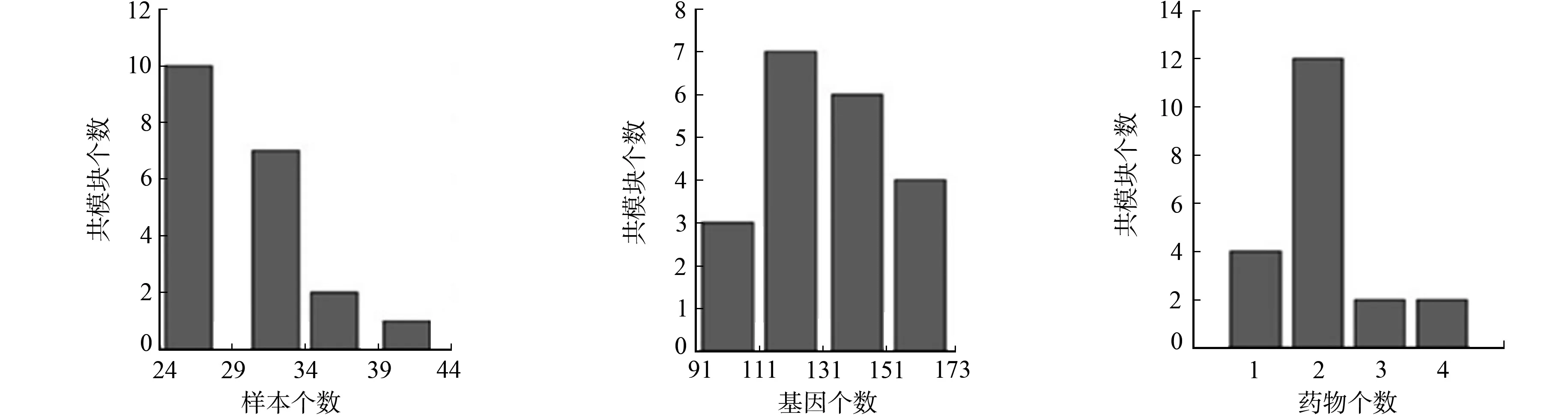
(a) 样本个数 (b) 基因个数 (c) 药物个数图2 SGDPLS模型识别共模块分布图Fig.2 Distribution map of SGDPLS model recognition common modules
为了解释文中识别出的共模块具有生物意义,利用R语言中的clusterProfiler包对识别出的基因模块进行GO生物功能项和KEGG通路富集分析.对于20个基因模块,有14个基因模块(70%)至少富集一种GO生物过程;有12个基因模块(60%)至少富集一种KEGG通路.例如,第2个共模块,样本模块包含28个样本,这些样本主要富集的癌症类型是小细胞肺癌和小细胞癌.第2个共模块生物项的富集结果,如图3所示.图3中:p表示富集分数经过Benjamini校正后的值.
基因模块包含107个基因,这些基因主要富集的GO生物过程是DNA构象变化、核染色体分离、姐妹染色体分离(图3(a)),这些过程都发生在细胞增殖过程中.因为姐妹染色体分离到子细胞的过程是不可逆的,检测点的缺失会导致染色体不稳定,所以,在细胞周期中,有丝分裂过程最易发生癌变[11].现代医学利用相关药物制止细胞中纺锤体的出现,从而抑制细胞有丝分裂的进行,使细胞分裂停留在G0阶段,利用该技术可以有效地遏制癌细胞的恶性增殖和扩散.另外,基因模块主要富集的KEGG通路为上皮细胞的细菌侵入、小细胞肺癌、细胞周期、焦点粘连、同源重组等细胞生化过程(图3(b)),这些生化过程对癌症的产生和转移起着很重要的作用.药物模块包含2个药物,即Docetaxel和RDEA119.Riichiroh等[12]指出,药物Docetaxel已在临床上被证实可以用于小细胞肺癌的治疗.药物RDEA119[13]是一种MEK抑制剂,MEK是一种磷酸化丝裂原活化蛋白激酶(MAPK)的激酶,MEK酶的靶标ERK具有高选择性,且可以驱动细胞增殖,从而实现抑制肿瘤细胞增殖并诱导细胞凋亡.研究表明,该药物对组织的选择性,可以降低其对中枢神经的毒副作用,且口服性的MEK抑制剂RDEA119已在临床上被开发应用,有效地推动了药代动力学的发展[14].

(a) BP (b) KEGG图3 第2个共模块生物项的富集结果Fig.3 Enrichment results of the second common module biological item
3 SGDPLS算法与SNPLS算法的比较
为了展示药物关联网络在SGDPLS算法中所起的重要作用,将SNPLS算法也用于同样的数据集上,分别计算2种算法识别出来的20个基因-药物共模块之间的皮尔逊相关系数,结果如图4所示.
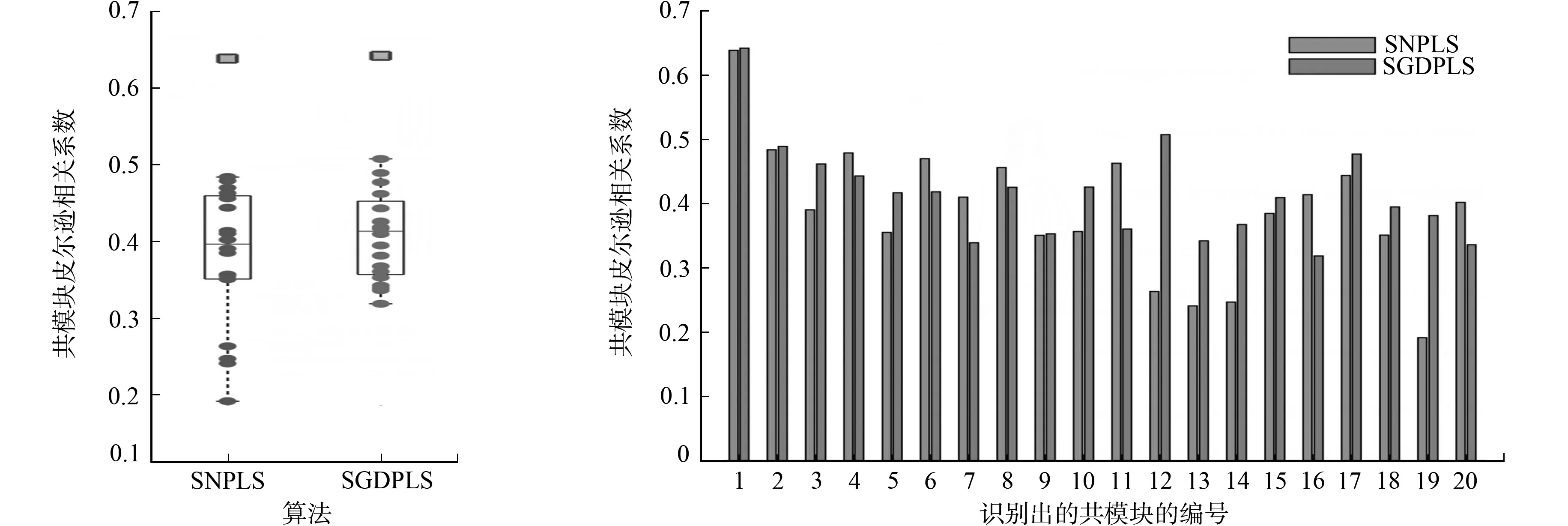
(a) 相关性分布箱线图 (b) 单个模块相关性柱状图 图4 两种算法共模块皮尔逊相关性比较Fig.4 Comparison of co-module pearson correlation between two algorithms
由图4可知:SGDPLS算法识别的大部分共模块(65%)比SNPLS算法具有更好的相关性,说明由药物二维化学结构构造的药物关联网络,在一定程度上,可以提高共模块中基因模块和药物模块数据模式的相似性,使共模块更具生物意义.
4 结束语
提出了SGDPLS算法,并深入挖掘多个基因和多个药物之间的对应关系,针对SGDPLS算法和SNPLS算法,独立识别出的20个基因-药物共模块,从共模块之间的皮尔逊相关系数和生物意义角度进行分析.结果表明:SGDPLS算法识别的共模块中,有65%比SNPLS算法识别的共模块具有更高的相关性.由此可见,药物关联网络信息的加入增强了SGDPLS算法识别模块的相关性,有助于发现潜在的药物靶标.同时,SGDPLS算法有望应用于其他领域中,进行高维数据的降维,如航天遥感数据、金融市场交易数据,用以发现数据的本质结构[15].
猜你喜欢
现代临床医学(2022年3期)2022-06-06
昆明医科大学学报(2021年12期)2021-12-30
新世纪智能(数学备考)(2021年9期)2021-11-24
好日子(2021年8期)2021-11-04
中学生数理化·高一版(2021年2期)2021-03-19
当代陕西(2019年15期)2019-09-02
领导决策信息(2018年16期)2018-09-27
海峡姐妹(2018年7期)2018-07-27
学苑创造·A版(2018年11期)2018-02-01
数学学习与研究(2017年3期)2017-03-09
