
看看这些罕见病有多罕见
2020-02-24 07:10安利
百科知识 2020年4期
安利
世界卫生组织将罕见病定义为患病人数占总人口的0.065%~0.1%之间的疾病或病变。国际确认的罕见病有五六千种,其中大多数是由于遗传缺陷所导致。为了引起公众对罕见病的关注以及对罕见病人的关爱,欧洲罕见病组织于2008年2月29日发起了第一届国际罕见病日。此后,世界多国将每年2月的最后一天定为国际罕见病日。有一些病症,如脊髓灰质炎、天花等曾经猖獗一时,但由于疫苗的广泛接种而变得极其罕见;下面这些病症,则不仅罕见,更多的时候人们连名字都没听到过。
1.哈钦森-吉尔福德早衰综合征
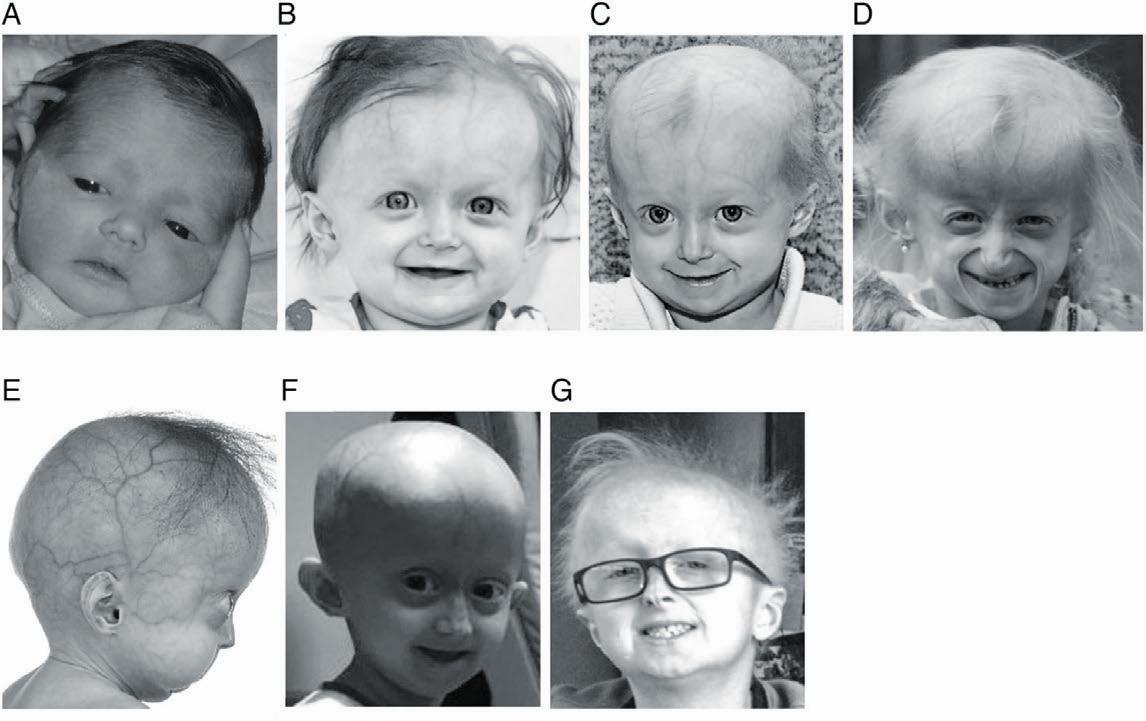
这种罕见病通常被称为儿童早衰症,大约每800万儿童中有一人患病。由于一种基因突变,导致儿童过早出现衰老迹象,如出现皱纹、秃顶,更危险的是出现动脉硬化,由此导致心脏病、中风的发生。大多数患有这种疾病的儿童只能活到13岁左右,很少有人能活过20岁。目前还没有治愈儿童早衰症的方法,只能减少并发症的发生。
2.高铁血红蛋白血症

这种疾病也被称为蓝皮病。先天性酶缺陷导致的高铁血红蛋白血症非常罕见,其特征是血液红细胞中高铁血红蛋白的含量超过正常值,使血液呈现巧克力色。正常血红蛋白含有的是二价铁,才能起到转运氧的作用,二价铁氧化成三价铁就使血红蛋白运氧能力减弱,导致患者更容易出现心脏异常、癫痫发作,甚至过早死亡。蓝皮病曾在美国肯塔基州偏远地区的一个家族中延续了200年。
3.周期性呕吐综合征
周期性呕吐综合征(CVS)这种罕见病以反复发作的严重恶心和呕吐为特征,一次发作可能持续数小时至数天,然后平复一段时间。这种发病和无病期的交替模式将 CVS与其他类似疾病区分开来。CVS的确切病因尚不清楚,对儿童的影响甚于成人,但成人发病持续时间可能更长。

4.库鲁病
库鲁病的罕见在于,它只现于巴布亚新几内亚东部高地的土著部落中,那里的土著人有食用已故亲人脏器的习俗。库鲁病由一种叫作朊病毒的蛋白质引起,朊病毒会导致异常的脑组织形成,而食用受感染者的大脑导致了疾病的传播。据称,这种100%致命的疾病在20世纪五六十年代曾造成1000多人死亡,但随着食人习俗的终结,库鲁病现已基本消失。

5. 菲尔德斯病
菲尔德斯病目前还没有正式的医学名称,暂以一对患病的双胞胎姐妹的姓氏命名。来自各地的医生研究了这对姐妹,认为她们患上的可能是一种神经肌肉疾病。它会引起每天多达百次的疼痛性肌肉痉挛,最终导致瘫痪并失去说话能力。

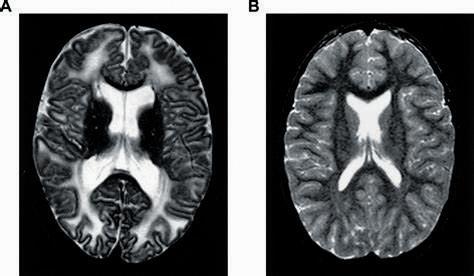
6. RPI缺乏癥
核糖-5磷酸异构酶缺乏症(RPI缺乏症)被认为是世界上最罕见的疾病,1984年,通过核磁共振成像在一名患者身上发现了一种脑白质疾病,通过DNA分析,最终于1999年确诊。但是,导致基因通路紊乱的分子机理尚不清楚。截至目前,全世界仅报道3例病例。
猜你喜欢
大自然探索(2022年5期)2022-07-11
今日农业(2021年15期)2021-11-26
上海工运(2020年8期)2020-12-14
科学(2020年3期)2020-11-26
世界科学(2020年11期)2020-11-24
今日农业(2019年13期)2019-08-12
中国兽医杂志(2018年4期)2018-01-25
工业设计(2016年4期)2016-05-04
中国中医药现代远程教育(2014年20期)2014-03-01
