
小儿肺朗格汉斯细胞增生症CT表现及其诊断价值
2020-02-17 13:16:46郑州大学附属儿童医院河南郑州450000
中国CT和MRI杂志 2020年1期
郑州大学附属儿童医院(河南 郑州 450000)
康乐乐 陈志平 时胜利
朗格汉斯细胞增生症(langerhans cell histiocytosis,LCH)是以器官内大量活化的朗格汉斯细胞增生、浸润为标志,一种少见的以单核-吞噬细胞系统特定的树突细胞增生为特点的疾病[1]。肺朗格汉斯细胞增生症(pulmonary langerhans cell histiocytosis,PLCH)是LCH患儿主要的死因之一[2],临床体征表现隐匿,加上小儿临床问诊的局限性,极易误诊、漏诊。现收集分析本院2010年4月~2017年11月经病理确诊的26例LCH患儿的影像资料,回顾分析不同病理时期PLCH的影像学表现,旨在提高对该病的影像学认识,协助临床准确诊断,制定针对性治疗方案。
1 资料与方法
1.1 一般资料 提取我院2010年4月至2017年11月26例经病理确诊为LCH患儿的病例资料,男18例,女8例,男女比例2.25:1,发病年龄1月6天~6岁2月不等,中位年龄1岁8月。病程3周至3年2月不等。15例有复诊记录,遵医嘱规范治疗后,5例肺部病变范围逐渐缩小好转;8例肺部病灶未再进展,但也无明显好转;2例病情难以控制,进展迅速,最终出现呼吸困难而死亡。余11例暂无复诊记录。本组患儿病例中肺部受累外,合并有皮肤受累者最多,有19例(73%),其次为骨受累,14例(53.8%),肝受累有12例(46%),淋巴结受累6例(23%),胸腺受累6例(23%),脾受累最少,仅5例(19%)。
1.2 仪器与方法 采用Philips Brillance 256排螺旋CT连续容积扫描。扫描范围由声门下至膈肌。扫描参数:100kV,76mA,螺距0.8,准直22mm,FOV:250mm×250mm,矩阵512×512。扫描完成后,采用Philips EBW工作站进行图像后处理,分别用不同窗宽、窗位观察。参数为:肺窗(W/C:1600/-600),纵隔窗(W/C:360/60)。
2 结 果
肺部CT表现多样,均有不同程度的间质性改变。影像学随不同的病理时期而变化:早期,富细胞期(6例):朗格汉斯细胞向外渗出,造成肺间质增厚,进而引起气道狭窄、气体滞留,影像学表现为肺野透过度不均,局限性或弥漫性磨玻璃影与小叶过度充气(图1),沿肺纹理走形逐渐出现网点状密度增高影且往往伴有小叶间隔的增厚(图2)。中期,增生期(16例):新生的肉芽肿侵犯邻近支气管及滋养血管,导致肺组织坏死,影像上表现为两肺结节影和(或)囊状气腔,散在分布,大小不等。结节与气腔的出现并无绝对的先后顺序,可先后出现或者两者共存。本组中11例结节与囊状气腔并存(图3);3例表现为弥漫性结节,两肺上叶居多,并无气腔形成;2例仅表现为肺部的局限性囊状气腔,肺内无结节存在(图6)。晚期,纤维化期(4例):肉芽肿破坏,纤维组织大量增生,肺大泡逐渐融合扩大,最终破溃造成气胸、纵隔积气(图8)。
3 讨 论
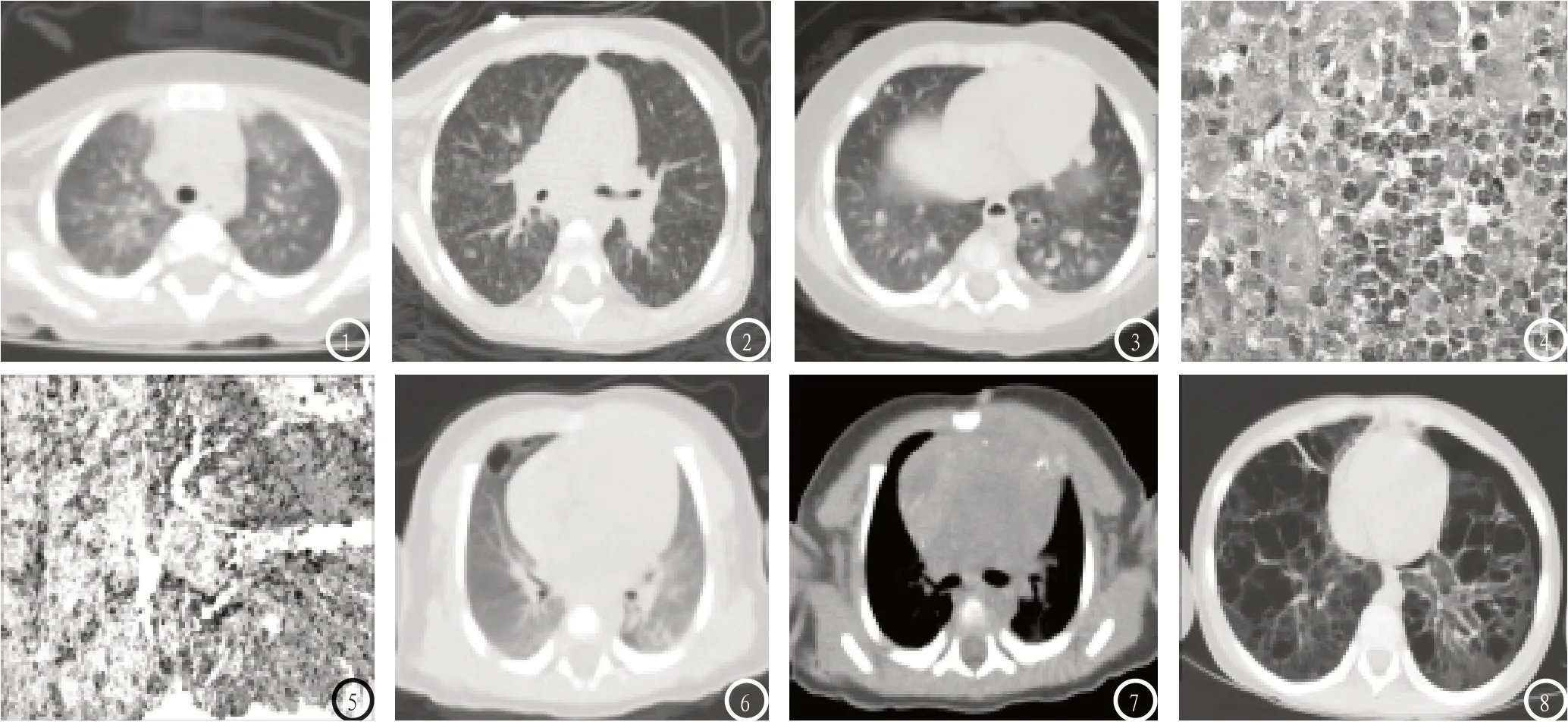
图1 女,7月22天,肺野透过度不均,局部小叶过度充气及磨玻璃样变,右肺上叶后段已出现边界模糊的结节影。图2 女,1岁8月,小叶间隔增厚,两肺多发网点状密度增高影,边界模糊,多沿支气管走形分布。图3-5 男4月21天,两肺多发结节,边界清晰,不对称、不均匀分布,部分结节出现中空,病理证实为朗格汉斯细胞增生症(嗜酸性肉芽肿),免疫组化:CD163(+),CD1a(+),CD56(-),CD68(+),CK(-),Langerin(+),S-100(+)。图6-7 男,1月6天,右肺见数个囊泡影,内有分隔,肺内并无结节,纵隔窗显示胸腺浸润,见多发条片状钙化及囊性变。图8 男,6岁2月,肺纹理扭曲,肺内弥漫性含气囊腔,形态不规则,部分融合,呈蜂窝状,同时合并有气胸及纵隔积气。
LCH是一类罕见的以组织细胞浸润为特征的疾病,以病变细胞中观察到Birbeck颗粒作为诊断的金标准,近年来大量研究证实Birbeck颗粒是由朗格素内化后形成[3],因此,近年来国际组织细胞协会指出,朗格素与Birbeck颗粒二者具备其一即可确诊[4]。LCH儿童发病率相对较低,约为3.5~7.0/100万[5],但病死率要远远高于成人,总体后遗症遗留率为30%~40%[6]。根据受累器官的不同,可分为单系统受累(single system,SS-LCH)和多系统受累(multi system,MS-LCH)。PLCH在成人多为肺部单独受累,儿童则为多系统受累伴肺侵害更为常见[7],本组病例中26例患儿均为多系统受累,除肺部受累外,合并有皮肤受累者最多,有19例(73%),其次为骨受累,有14例(53.8%)。PLCH的临床体征比较隐匿,从无明显呼吸道症状到呼吸困难表现多样,本组中有22例(84.6%)患儿存在不同程度的呼吸道症状,其余4例(15.3%)并无肺部体征,后行肺部CT筛查时发现异常。PLCH随着病情的进展可出现3种病理状态:富细胞期、增生期、纤维化期[8]。富细胞期以LC、巨噬细胞等向外渗出为特征,随着渗出的增多,范围的扩大而形成结节。增生期:结节数目不断增加,空洞型肉芽肿形成,并出现纤维瘢痕。纤维化期:肉芽肿破坏,空洞形成,增生的纤维组织围绕在囊腔周围,形成蜂窝肺,同时肺血管壁受浸润后中膜肥大、管腔变窄,最终造成顽固性肺动脉高压[9]。本组病例中肺部CT表现与病理表现相符,其中结节与囊泡的出现几率最高,有16例,且以两肺中上肺野居多,这与相关报道[10]相符。
PLCH的病因及发病机制尚不明确,细胞因子的促发可能会诱导朗格汉斯细胞在器官内聚集[11]。有研究发现LCH患儿中所有的单核细胞亚群均分泌IL-17A,且IL-17A的表达水平与LCH疾病活性相关[12]。另TNF-α、IL-1、IL-4等细胞因子参与PLCH病变的也有报道。除了细胞因子促发,吸烟是PLCH的另一个重要致病因素。但这些都处于假说阶段,并未得到定论。
PLCH在不同病理时期的影像学表现也不同:早期可有磨玻璃影、小叶过度充气征象或者沿支气管分布的网点影;中期以结节影和囊状空腔为主要表现;晚期纤维组织大量增生、含气囊腔相互融合,呈蜂窝样改变,并出现气胸、纵膈积气及肺动脉高压等病晚期征象。当部分病例影像学不典型时,可通过肺泡灌洗液免疫组化结果来帮助诊断。本病有时与其他一些如肺嗜酸细胞增多症,特发性肺含铁血黄素沉积症,白血病肺浸润等表现类似,几者之间都有不同程度的肺间质改变,但PLCH更多伴有囊泡、结节影及肺外脏器的损害,结合相关病史及其他实验室检查更有助于鉴别。
总之,PLCH的发病机制不明,常规实验室检查多无异常,临床症状也表现多样,容易出现误诊、漏诊。有研究数据显示LCH患者早期应用激素、抗生素、放化疗等方法后病情大多都能得到有效缓解[13],所以PLCH的及时诊断就显得尤为重要。除了组织活检,CT在PLCH的诊断中也不可或缺。若临床症状不典型,而肺部CT有间质性病变,甚至出现结节和气囊,且以两肺上叶居多时,应建议进一步检查排除PLCH,尽量让患儿及时确诊治疗,让病情得到有效控制。
猜你喜欢
润滑与密封(2023年5期)2023-05-25 02:37:06
中国科技纵横(2023年4期)2023-05-18 13:07:22
家教世界·创新阅读(2022年4期)2022-05-07 20:35:51
情感读本·道德篇(2021年7期)2021-12-14 12:39:44
风流一代·经典文摘(2021年10期)2021-10-25 09:38:21
制造技术与机床(2021年5期)2021-06-18 03:08:26
国际放射医学核医学杂志(2020年2期)2020-05-30 12:40:00
爆炸与冲击(2018年6期)2018-10-16 08:53:10
小品文选刊(2016年1期)2016-02-12 03:56:50
教育与职业(2014年28期)2014-01-19 01:46:36
