
纳米结晶纤维素表面修饰聚酰亚胺纤维及其润湿功能性
2020-01-15 08:29:44党洪洋张国亮龙柱王士华李志强胡爱林郭帅吕文志
化工进展 2020年1期
党洪洋,张国亮,龙柱,,王士华,李志强,胡爱林,郭帅,吕文志
(1江南大学生态纺织教育部重点实验室,江苏无锡214122;2连云港纤维新材料研究院有限公司,江苏连云港222002;3连云港市工业投资集团有限公司,江苏连云港222002;4黔南民族师范学院,贵州都匀,558000)
聚酰亚胺(PI)纤维因高强度、高模量、耐高温、绝缘性能好等优点,被广泛应用于军事、航空航天、汽车等领域[1-3]。以PI 纤维作为基材的复合材料需要纤维具有良好的界面性能及与其他材料较好的相容性,这些性能与纤维的润湿性关联紧密,由PI 纤维到PI 纤维原纸的湿法抄造过程要求PI 纤维与水有更好的相容性以提高纤维的分散性,进而得到匀度更高的纸基材料。但PI 纤维分子结构中含有稳定的酰亚胺基团,使纤维表面钝化,没有极性基团,且大分子排列规整,结晶度高,所以水分子通过PI 纤维表面进入纤维内部的阻力很大。同时经纺丝而来的PI纤维表面光滑且没有微细纤维,使得纤维的浸润性较差,这极大地阻碍了纤维在液相中的分散性,限制了纤维的进一步推广应用[4-5]。因此,对PI纤维进行表面处理,克服其表面缺陷,对增强PI纤维润湿性能极为重要。
近些年来,对PI 纤维改性一直是国内外研究工作的重点和热点,纤维的表面改性也是科研工作和工业应用的主要手段之一[6-7]。杨冰磊等[8]采用辉光低温等离子体对聚酰亚胺纤维进行表面处理,利用等离子体中被激发的活性粒子在聚酰亚胺纤维表面发生刻蚀、粗化作用,使纤维表面产生接枝、降解、交联等反应而出现微刻蚀,活性官能团增加,纤维吸湿性能提高。魏宁[9]采用弱碱性的乙二胺溶液对聚酰亚胺纤维进行处理,在纤维表面引入了活性基团,处理之后的聚酰亚胺纤维所成的纸页接触角降低了42.4%,证明了纤维润湿性能得到改善。但这些方法对纤维自身强度有所影响,阻碍了纤维材料更加广泛的应用。Chen等[10]采用连续浸渍涂覆法在碳纤维表面沉积一层聚多巴胺薄膜,使得碳纤维由两憎物质转变为亲水性物质,从而增强了纤维与聚合物基质的界面黏结强度。然而,这种在纤维表面易形成非共价键的作用力具有一定的不稳定性和时效性,同时易造成材料性能的损失。
纳米纤维素作为一种天然高分子材料,具有高比表面积、高抗拉强度、高亲水性和高活性等特性,同时,对于纤维材料的性能有一定的增强效果[11-13]。如图1 所示,通过表面改性在PI 纤维表面引入的纳米结晶纤维素(CNC)可以达到两种材料的优势互补作用,既增加了PI 纤维的表面极性,又改善了PI 纤维的润湿性能,且CNC 独特的三维网状交联结构对经碱处理后纤维性能的下降有部分补强作用。
本文拟在复合路易斯酸催化剂和BTCA交联剂催化引发下,使CNC 与碱处理后的PI 短切纤维发生酯化反应,并优化接枝反应的工艺条件,以提高PI 纤维的润湿功能性及其在水相中的分散性,期望利用CNC 对PI 纤维表面的接枝改性反应,为PI纤维的润湿性改善提供绿色环保的技术支持。
1 实验材料及方法
1.1 主要材料和仪器
PI 短切纤维,江苏奥神新材料股份有限公司,主要技术参数见表1;纳米结晶纤维素(TOCNPS150,CNC),粒径150~200nm,天津市木精灵生物 科 技 有 限 公 司; 乙 醇(C2H6O)、 丙 酮(CH3COCH3)、氢氧化钠(NaOH)、盐酸(HCl)及无水氯化铝(AlCl3),国药集团化学试剂有限公司;对甲苯磺酸(PTS),分析纯,上海埃彼化学试剂有限公司;1,2,3,4-丁烷四羧酸(BTCA),分析纯,上海麦克林生化科技有限公司。

表1 聚酰亚胺短切纤维的主要技术参数

图1 PI纤维表面接枝CNC机理图
立式标准纤维疏解器,PL28-2 型,咸阳泰思特试验设备有限公司;纸样抄片器,ZQJ1-B-Ⅱ型,陕西科技大学机械厂;傅里叶变换红外光谱仪(FTIR),Nicolet is10型,美国赛默飞世尔公司;X射线光电子能谱仪(XPS),Thermo ESCALAB 250XI型,美国赛默飞世尔公司;扫描电子显微镜(SEM),su1510型,日本日立株式会社;热重分析仪(TG),Q500 型,美国TA 仪器公司;光学接触角表面分析仪,DSA100型,德国LAUDA Scientific公司;多孔材料孔径分析仪,CFP-1100A 型,美国PMI公司。
1.2 实验方法
1.2.1 聚酰亚胺短切纤维前处理
以质量分数为50%的乙醇对PI 纤维进行超声清洗2h,除去PI 纤维表面纺丝油剂及其他附着杂质,再以去离子水多次清洗,取出后置于60℃烘箱中烘干4h待用。
1.2.2 聚酰亚胺短切纤维碱处理及酸质子化
配制500mL 质量分数为5%的NaOH 溶液置于磁力搅拌水浴锅中充分溶解,升温至60℃加入PI纤维,密封碱处理1h,将处理后的纤维用去离子水多次清洗,洗净纤维表面残余碱液,取出后置于60℃烘箱中烘干4h待用。
配制500mL 质量分数为2%的HCl 溶液,加入碱处理后的PI纤维,于磁力搅拌器中密封处理1h,将处理后的纤维用去离子水多次清洗,洗净纤维表面残余酸液,取出后置于60℃烘箱中烘干4h待用。
1.2.3 正交试验设计
根据初步的试验探究,为提高实验效率,不考虑各因素之间的相互作用,在本实验条件下,确定了影响PI 纤维表面接枝的主要因素有CNC 浓度(A)、BTCA含量(B)、反应时间(C)和反应温度(D)。对实验进行了四因素二水平的正交设计,以PI 纤维成纸关于去离子水的接触角(CA)为评价标准,采用极差分析法分析实验结果。
通过表2实验优化得到最佳的反应参数,并以优化后的组合方案进行表面接枝实验。
1.2.4 纳米结晶纤维素表面接枝聚酰亚胺短切纤维
将处理后的PI 纤维与250mL 一定浓度的CNC溶液加入到三口烧瓶中,升温至40℃;以去离子水为溶液,质量分数为0.5%AlCl3和质量分数为0.3%PTS复合的路易斯酸作为催化剂,以一定浓度的BTCA作为交联剂,通过超声处理充分溶解混合均匀后加入到烧瓶中,反应混合物通过氮气保护在一定温度下连续磁力搅拌反应一定时间。通过真空抽滤法取出反应产物,并分别用丙酮和去离子水对其进行超声清洗1h,于60℃烘箱中干燥4h 得到接枝产品。

表2 正交实验因素水平表
1.2.5 聚酰亚胺短切纤维成纸的制备
按照聚酰亚胺短切纤维和芳纶浆粕为7∶3 的比例置于立式标准纤维疏解器中疏解15000转,后于纸样抄片器抄取定量为60g/m2的纸页。
1.3 性能测试及表征
1.3.1 红外光谱测试
采用傅里叶变换红外光谱仪对处理前后PI 纤维的化学结构进行表征。扫描次数为16,扫描范围为400~4000cm-1,分辨率4cm-1。
1.3.2 X射线光电子能谱测试
采用X射线光电子能谱仪测试处理前后纤维的表面元素组成及官能团变化。以位于284.6eV处的C1s 信号为基准进行曲线校准,并对C1s 峰图谱的曲线采用最小二乘法进行拟合。
1.3.3 扫描电子显微镜
采用扫描电子显微镜观察处理前后纤维表面的外观形貌,并进行比较分析。用含银导电胶作为基面,喷金处理后进行测试,所用电压为20kV。
1.3.4 热重测试
采用热重分析仪表征处理前后PI 纤维的热性能。测试时,取样1~3mg 置于坩埚中,在氮气保护下,以5℃/min的速度从室温升温至800℃。
1.3.5 润湿性测试
取经CNC 处理前后的PI 纤维成纸(3cm×3cm),保持表面平整,置于光学接触角表面分析仪的载物台上,通过针头缓慢挤出浸润液滴于纸页上,计时2s,记录所测得接触角。实验中选择的浸润液为去离子水(γl=72.8mJ/m2,=21.8mJ/m2,=51.0mJ/m2)和无水乙醇(γl=48.0mJ/m2,=23.0 mJ/m2,=19.0mJ/m2)。
利用PI 纤维成纸对浸润液的接触角数据及根据式(1)和式(2)计算出PI 纤维成纸表面自由能中非极性分量和极性分量的值,从而得到PI 纤维成纸的表面自由能γf。

式中,θ为PI 纤维对测试液的接触角,(°);γf为表面自由能,mJ/m2;为表面自由能中的非极性分量,mJ/m2;为表面自由能中的极性分量,mJ/m2;下标l代表测试液;下标f代表纤维。

表3 测试液的表面能(20℃)
1.3.6 分散性能测试
将纤维与水按照3‰的质量比加入量筒中,静置12h,观察纤维悬浮液的沉降情况,根据式(3)计算纤维的分散度[14]。

式中,v0为开始沉降时纤维悬浮液的总高度,mL;v1为静置12h 后悬浮液中上清液层的高度,mL;f为纤维分散度,%。
1.3.7 聚酰亚胺成纸纤维分布状况
在PI 纤维成纸上裁取3cm×3cm 的样品,使用多孔材料孔径分析仪检测纤维纸页的孔径分布状况。
2 结果与讨论
2.1 CNC表面接枝PI短切纤维工艺优化
CNC 表面接枝PI 短切纤维的正交试验结果如表4所示。
表4 给出了L8(24)的正交实验及CA 的测试结果。从表4 可以看出,各因素对CA 影响程度的排序为CNC 浓度>反应温度>反应时间>BTCA 质量分数。其中,在本实验条件下,最佳的CNC 质量分数为3.12%,最佳温度为70℃,最佳反应时间为5h,最佳BTCA 质量分数为0.4%。且PI 纤维成纸关于去离子水接触角都得到了降低,最大降低了14.6°。
2.2 纤维表面元素及官能团变化
图2 是经碱处理和CNC 处理前后PI 纤维的红外光谱图,从谱图(a)中可以看出,未处理的PI 纤维在1775cm-1、1714cm-1的特征峰是酰亚胺基团中的羰基()对称振动,1367cm-1的吸收峰对应于酰亚胺基团中碳氮键()伸展振动,1307cm-1处为酰亚胺基团中碳氮键()伸缩振动引起的吸收谱带,即酰胺Ⅲ带,在1496cm-1处存在苯环结构的振动峰,719cm-1和820cm-1处的吸收峰证明苯环发生了间位取代[15]。
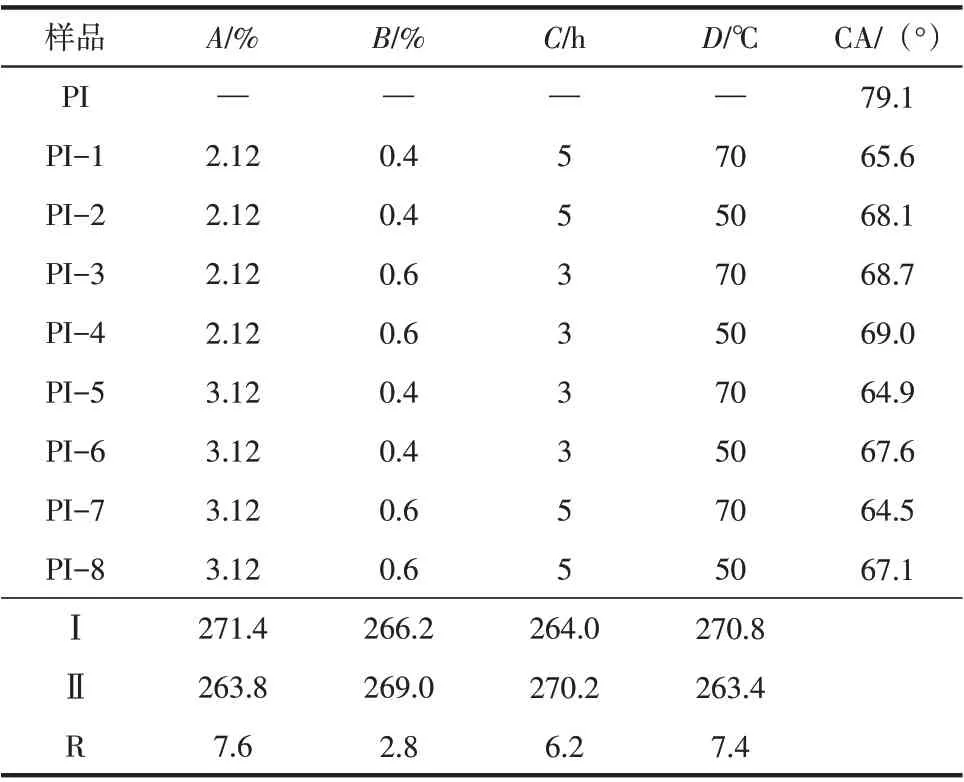
表4 正交试验结果

图2 CNC处理前后纤维的红外光谱图
经碱处理后,PI 纤维红外谱图发生变化,1775cm-1、1714cm-1、1367cm-1等处的吸收峰明显减弱,而在1647cm-1、1547cm-1处出现新的特征吸收峰,分别对应仲酰胺基团()中的碳氧双键()伸缩振动和氮氢键()弯曲振动,说明碱处理后的PI 纤维表面暴露出了仲酰胺基团和羧酸基团,有利于下一步酯化反应的进行[16]。而后,经CNC 处理后的PI 纤维红外谱图中,位于1714cm-1、1307cm-1等处的吸收峰明显减弱甚至消失,同时在3330cm-1的吸收峰出现了CNC 分子链上羟基( OH) 的伸缩振动,2915cm-1、2847cm-1分别为纳米纤维素上碳氢键(C H)的不对称伸缩振动和对称伸缩振动峰,初步说明处理后的PI纤维表面存在CNC。
图3与图4分别是CNC 处理前后PI纤维的XPS全扫描图和C1s 解析谱图。由图3可知,处理前后的PI纤维都具有相同的C1s、N1s、O1s峰,这表明纤维表面由相同的元素组成,但处理后PI 纤维的O1s 峰的强度显著增强,而C1s 和N1s 峰强降低,这与红外谱图的结果相吻合。
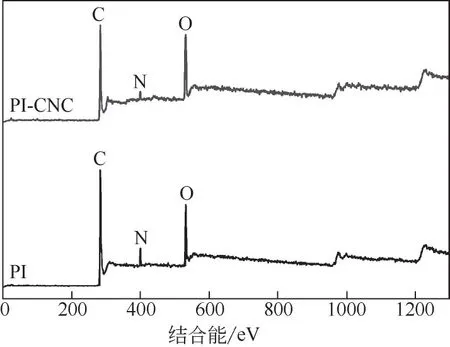
图3 CNC处理前后纤维的XPS全扫描图
通过XPS Peak Fit分峰软件对C1s进行分峰拟合,定量计算出纤维表面C1s、N1s、O1s的质量分数并列于表5中。由表5可得出,经处理后O1s的质量分数明显升高,由15.98%增加到19.74%;而C1s和N1s含量明显降低,由78.02%和6.00%分别降低到75.39%和4.87%,氧元素的增加主要归因于在纤维表面接枝的纳米结晶纤维素(CNC),而CNC中含有大量包括羟基在内的含氧基团。未处理的PI纤维的C1s经拟合后可分出3个峰,284.5eV处的结合能对应的是C C,285.6eV 处的结合能对应的是C N,287.9eV处的结合能对应的是,这均反映了PI纤维的本体结构;而经CNC处理后的PI纤维的C1s在289.4eV处出现了新峰,对应的是COOR,这是因为处理后纤维表面接枝了一定量的CNC[17-18]。

图4 CNC处理前后纤维的C1s解析图谱

表5 CNC处理前后纤维表面元素含量的变化
综上所述,可以得出经CNC 处理后在PI 纤维表面接枝上了部分CNC,引入了亲水性基团羟基,提高了纤维表面的活性,进一步促进了纤维与极性溶剂的相容性,纤维的润湿性得到增强。
2.3 纤维表面形貌变化
图5 为PI、PI-COOH 和PI-CNC 纤维表面形貌图。从图中可以看出未经处理的PI 纤维表面平整光滑,无突起和刻蚀,呈圆柱状。经碱处理及酸质子化后的PI纤维表面发生了物理和化学两个过程。物理过程是经刻蚀后纤维表面而出现的刻槽和凸点,化学过程是碱液渗入纤维内部无定型区及环结晶区进而造成PI 大分子中主链酰胺键发生水解,使得纤维表层发生腐蚀作用。
从图5(c)经CNC 处理后的PI 纤维表面可以看出,碱刻蚀产生的凹槽缺陷被纳米纤维素微粒填平,且伴有纤维表面纳米结晶纤维素的聚集,使得大量的“花朵”或“花瓣”状的团聚体出现在纤维表面,以至于相互之间发生了物理缠结或是化学交联,从而在纤维表面形成了一层不均匀的CNC 结构,增加了PI 纤维表面的粗糙度,同时由于CNC含有丰富的羟基,使得纤维表面极性增加,进一步提高了纤维的亲水性能。
2.4 纤维热学性能变化
图6 为经碱处理和CNC 处理前后纤维的TG 和DTG 曲线,可以看出,未处理的PI 纤维初始降解温度在500℃左右,PI 纤维在500~760℃存在一段单失重峰。经碱液处理后得到的PI COOH纤维表面分子链发生降解,分解温度向低温区域偏移,分别在343~411℃和411~644℃出现两组热失重峰,343~411℃的范围内的失重是碱处理后的纤维大分子残片与暴露出的纤维分叉结构热分解的结果[19]。
经纳米结晶纤维素处理后得到的PI-CNC 纤维在115~200℃和419~687℃出现两组失重峰,而经碱处理后得到的PI-COOH 纤维的位于343~411℃的失重峰消失,在120~210℃内的失重PI-CNC 纤维表面所形成的的网状结构发生分解的结果[20],经CNC 表面修饰处理后得到的PI-CNC 纤维表面生成CNC 和PI 纤维的缠结及纳米颗粒在纤维表面形成的不规则包覆结构,补偿了碱处理引发的非晶区域增加而造成的热分解温度峰值降低和分解温度区域偏移现象,除了表面的CNC 交联包覆层失重温区较低外,纤维的热稳定程度较碱处理后有所提高,说明补偿了部分PI纤维损失的热性能。

图5 CNC处理前后纤维的SEM图

图6 CNC处理前后纤维的TG和DTG图
2.5 纤维亲水性能变化
在相同工艺条件的前提下,经CNC 处理前后的PI短切纤维在水体中的分散状态如图7所示。未处理的PI 短切纤维在水溶液中分布不均,纤维间相互缠绕呈现“棉球状”或“絮状”,分散状态差;而经CNC 处理后的纤维分散体系中PI 短切纤维基本呈单丝状态,分散较为均匀,呈悬浮状态,并与水体表现出很好的相容性,宏观上说明了纤维表面润湿性能得到增强。由表6可知,由于经CNC处理后的纤维表面引入了极性基团,且CNC 晶粒尺寸较小,其自身形成交联结构团簇于经碱处理后的纤维表面,获得纳米尺寸的粗糙表面,使得纤维比表面积和表面粗糙度增大,亲水性增强,静置12h后纤维的分散度提高了45%。

图7 纤维在水体中的分散状态图

图8 CNC处理前后纤维原纸的接触角
接触角大小直接反映液体介质对PI 短切纤维的润湿性。如图8所示,采用光学接触角测试仪测试PI 纤维成纸关于去离子水和乙醇两种润湿液的接触角,同时根据Owens-Wendt 方程计算出PI 纤维成纸表面自由能的非极性分量()、极性分量()及表面自由能(γ)f,结果列于表6[21]。可以看出PI 纤维成纸对去离子水还是乙醇的接触角都相对较大,说明PI 纤维对强极性溶剂和弱极性溶剂润湿性都较差。相对的,PI-CNC 纤维成纸对以上两种溶剂的接触角都有了明显减少,纤维的润湿性得到提高,表面粗糙度的增加及极性官能基团的引入是纤维表面润湿性能提高的主要原因。同时,由于引入的官能基团是亲水性极强的羟基,因此PICNC 纤维成纸对去离子水的浸润性提高程度明显大于其对乙醇的浸润性提高程度,PI-CNC 纤维成纸对去离子水润湿液的接触角降低了14.9°,而对乙醇润湿液的接触角只降低了4.8°。

表6 纤维的分散度及纤维成纸的接触角和表面能(20℃)
表面自由能(γ)f由非极性分量()和极性分量()组成,CNC 处理前后PI 纤维成纸的表面自由能数据如表6 所示。从中可以看出,PICNC纤维成纸的表面自由能相对于PI纤维成纸得到了明显提高,PI-CNC 纤维成纸的表面自由能为38.20mJ/m2,约为PI纤维成纸表面自由能的1.3倍。一方面是因为在纤维表面引入了CNC 后,能够促进其克服液体的表面张力,使液体更易在纤维表面铺展开来。另一方面,与PI相比,PI-CNC 表面自由能中的极性分量()显著增加,非极性分量()明显减少,这说明PI-CNC表面自由能的增大主要源于极性分量()的贡献。同时说明了表面粗糙度的增大和极性活性基团的增多都有利于接枝处理后的纤维克服浸润液体的表面张力,且使浸润液体在接枝处理后的纤维表面上更易铺展和渗透。
2.6 聚酰亚胺纤维成纸中纤维分布变化
从图9(a)纤维成纸4 个样点图及图10(a)纸页的孔径分布图可以看出,未经处理的纤维制得的纸页中纤维分散性差,纤维间所成孔隙尺寸各异,纸页均一性差,约53.80%的孔径分布于10~50μm 间,110~150μm 范围内的占比约19.00%,其余尺寸范围内的孔径均存在,这种纤维分布的不均匀性致使纤维间发生缠结,纤维束较多,纤维层数不均一,致使纸页间应力传递不佳,影响了成纸的匀度及力学性能。

图9 纤维成纸中纤维分布情况
相对而言,PI-CNC 纤维成纸纤维间形成的孔洞较密集,孔隙分布相对较为均匀。由图10(b)看出处于110~150μm 的孔径占比约为5.56%,相对降低了13.44%,10~50μm 间的孔径分布比率达到77.84%, 相对增加了约24.04%,同时可看出孔隙间相互连通程度降低,孔隙尺寸变小,数量增多,在微观上说明了分散性得到了改善,与纤维分散度增加的结论相互支持,这主要是由于纤维的亲水性增加,在湿法抄造过程中,更易分散为单根纤维进而形成孔径均一并具有三维网状结构的纸页,有利于实现纸基材料良好的应力传递,改善了成纸匀度和力学性能。

图10 纤维成纸孔径分布
3 结论
(1)在复合路易斯酸及交联剂作用下,通过纳米结晶纤维素对聚酰亚胺纤维表面进行了修饰,使得PI 纤维的表面由原先的光滑结构转变为由纤维与CNC 形成的交联结构及CNC 纳米微粒之间形成的不规则粗糙包覆结构,极性基团的引入及纤维表面粗糙度的增加显著改善了纤维的润湿功能性及其在水体中的分散性,纸页匀度也得到了提升。
(2)讨论并优化了CNC 表面改性PI 短切纤维的工艺条件,并以纤维成纸对去离子水的接触角(CA)作为判定标准,本实验中最优的反应条件是:CNC 质量分数3.12%,BTCA 质量分数0.4%,反应温度70℃,反应时间5h。
(3)通过改性增强了PI 纤维的表面活性,同时补偿了部分碱处理后纤维损失的耐热性能,本方法可用于在尖端工业推进受制于其表面性能缺陷的PI纤维的发展。
猜你喜欢
中国造纸学报(2022年1期)2022-05-13 03:57:54
小资CHIC!ELEGANCE(2021年41期)2021-11-08 09:43:22
纺织科技进展(2021年5期)2021-07-22 08:41:36
原子与分子物理学报(2021年1期)2021-03-29 07:28:30
现代塑料加工应用(2021年5期)2021-02-28 08:19:20
实用口腔医学杂志(2017年6期)2017-09-19 02:51:14
西南石油大学学报(自然科学版)(2016年6期)2017-01-15 14:14:19
中国石油大学学报(自然科学版)(2015年2期)2015-11-10 06:08:25
造纸化学品(2015年2期)2015-11-04 06:03:22
中国塑料(2015年2期)2015-10-14 05:34:10
