
小花棘豆Alternaria oxytropis内生真菌酵母氨酸还原酶基因的Southern blot检测分析
2020-01-06 07:29珠丽卢萍席领军高峰李玉玲姜凯
生物化工 2019年6期
珠丽,卢萍,席领军,高峰,李玉玲,姜凯
(内蒙古师范大学生命科学与技术学院,内蒙古呼和浩特 010022)
小花棘豆(Oxytropis glabra DC.)是豆科棘豆属多年生草本植物,许多植株内含生物碱苦马豆素(swainsonine,SW),国际上将含SW的棘豆属(Oxytropis)、黄芪属(Astragalus)等有毒植物统称为疯草,而SW是引起动物疯草病的唯一毒素[1]。SW阳离子半椅式构象与甘露糖阳离子构象类似,可与甘露糖苷竞争性结合抑制α-甘露糖苷酶Ⅰ和高尔基体α-甘露糖苷酶Ⅱ的活性[2],造成细胞功能紊乱,使牲畜出现中毒症状,严重时致死。通过对小花棘豆的研究发现,凡能够分离或检测到Alternaria oxytropis内生真菌的植株均含SW,且体外培养该内生真菌合成了SW,从而提出小花棘豆的SW毒性由该内生真菌引起。
疯草内生真菌酵母氨酸还原酶基因(saccharopine reductase gene,sac)编码酵母氨酸还原酶[3-4]催化赖氨酸转化为酵母氨酸后,生成的α-氨基己二酸半醛是内生真菌合成SW的一个重要前体[5]。前期实验中克隆到A.oxytropis的sac cDNA(KJ944635)和基因(KY052048),获得sac缺失突变体M1,发现酵母氨酸还原酶促进真菌SW的合成。
Southern blot由Southern[6]于1975年创建,是分析基因结构和检测特定DNA片段的经典技术,可检测外源目的基因是否已转入并整合到生物染色DNA上,同时可对外源片段拷贝数进行分析。Southern blot中使用的标记探针有放射性同位素和非放射性两类,其中放射性探针灵敏度较高,但半衰期短,且有放射性辐射;非放射性标记又分为生物素标记和地高辛标记,非同位素标记探针可避免放射性危害,且由于生物体内没有地高辛,可更好地消除内源背景,应用效果更好[7]。
Southern blot操作程序繁琐,对基因组DNA的提取、纯化、酶切等均有较高要求,要获得理想杂交结果需反复摸索,虽可使用相关试剂盒,但需根据具体情况优化杂交方案。本研究利用PCR制备地高辛(DIG)标记探针,并对Southern blot方法进行了探索和优化。
1 材料与方法
1.1 材料
1.1.1 实验菌种
供试菌种:小花棘豆A.oxytropis内生真菌野生株OW7.8和sac缺失突变株M1由本实验室提供。
1.1.2 主要试剂
质粒提取试剂盒、琼脂糖凝胶 DNA 回收试剂盒,购自Axygen;真菌基因DNA 提取试剂盒、核酸共沉剂、限制性酶、6×Loading Buffer、DL2000 DNA Marker,购自Takara;核酸染料购自Bio TAQ;DIG标记试剂盒、Hyb高效杂交液,购自北京美莱博医学科技;氨苄青霉素、潮霉素 B,购自sigma;其他试剂均为国产分析纯试剂。
1.1.3 主要仪器
超净工作台ZHJH-1109 型,上海智城分析仪器制造有限公司生产;真菌培养箱MGC-350BP-2型,上海一恒科技有限公司生产;冷冻离心机3-18K/D-37520 型,德国 SIGMA生产;超微量测浓仪Q5000型,美国 Quawell生产;PCR 扩增仪050-810Tgradient48 型,德国Biometra-grandien生产;全自动高压灭菌锅HVE-50 型,日本 HIRAYAMA生产;电泳仪BG-Power 600型,北京百晶生物技术有限公司生产;凝胶成像系统BG-gds AUTO,北京百晶生物技术有限公司生产;超声波清洗器PS-20,洁康超声波设备有限公司生产;Hybridization oven OV4000,德国耶拿分析仪器公司生产;隔水培养箱GH6000,天津泰斯特仪器生产。
1.2 方法
1.2.1 PCR法制备DIG标记探针
根据内生真菌sac中间序列设计特异性引物DSF/DSR和SEMF/SEMR,以pTOPO-sac1(含内生真菌sac)为模板制备野生型探针。PCR反应体系为:pTOPO-sac11.0 μg,10×PCR Buffer 2.0 μL,DigdUTP 1.0 μL,rTaq酶1.0 μL,上下游引物各1.0 μL(10 μmol/L),ddH2O补足至20 μL。引物DSF/DSR的PCR反应条件:95 ℃ 5 min;95 ℃ 30 s; 57 ℃30 s;72 ℃ 1 min,30次循环;72 ℃ 10 min。引物SEMF/SEMR的PCR反应条件:95 ℃ 5 min,(95 ℃30s,62 ℃ 30s,72 ℃ 30s)×30,72 ℃ 10 min。
根据pCB1003(含hph)质粒上的hph序列设计特异性引物HGF/YGR,以质粒为模板制备突变型探针。PCR反应体系除模板其余与上面体系相同。PCR反应条件与DSF/DSR的相同。引物序列见表1。
在做标记反应的同时,做一个相同体系非标记PCR。反应结束后取标记和非标记PCR的扩增产物进行电泳检测(1%琼脂糖),剩余探针保存于-20 ℃,用于后续实验。
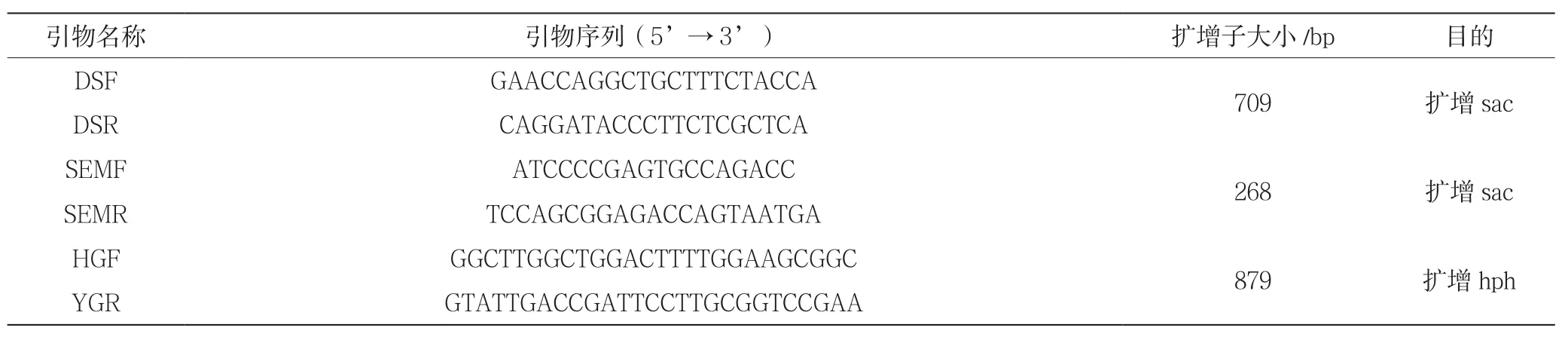
表1 PCR引物序列
1.2.2 基因组DNA提取、酶切与转膜
(1)真菌基因组DNA提取
将OW7.8和M1接种于PDA培养基上,M1培养基含潮霉素B(终浓度为50 μg/mL),接种后置于25 ℃培养箱中培养14 d。
刮取50 mg菌丝体放入预冷研钵中,加液氮迅速研磨至粉末,后置离心管中,加500 μL 4×CTAB提取液(含1% β-巯基乙醇)、4 μL RNase(100 mg /mL)充分摇匀,65 ℃水浴保温40 min,再加等体积酚/氯仿/异戊醇(25∶24∶1)混匀,4 ℃12000r/min离心5 min。在上清液中加2.5倍体积预冷无水乙醇,4℃12000r/min离心15 min,弃上清液。70%乙醇离心洗涤两次,4℃12000r/min离心5 min,弃上清液,80℃烘10 min,加100μL ddH2O,37 ℃水浴1 min,测定DNA浓度及OD值,取5μL进行电泳检测。
(2)基因组DNA限制酶切反应
选择限制性内切酶Eco RⅤ、Pst Ⅰ、Pci Ⅰ、PvuⅡ进行酶切反应,总体积50 μL,反应体系见表2和表3。基因组DNA终浓度调至20 ng,于37 ℃酶切12 h,视情况延长反应时间至完全酶切,65 ℃保温10 min,停止反应。使用核酸共沉剂进行酶切产物的纯化和浓缩,用琼脂糖凝胶电泳检测(0.8%)。

表2 OW7.8基因组DNA酶切反应体系
(3)转膜和固定
向上毛细管转移20 h以上。转膜结束后,用凝胶成像系统检测效果。取膜,切右上角标示方向。将尼龙膜浸入6×SSC溶液中5 min,80 ℃烘烤2 h,进行膜固定,固定后用于杂交。

表3 突变株株基因组DNA酶切反应体系
1.2.3 Southern blot
加Hyb高效杂交液50 ℃预杂交3 h(15 r/min)以封闭非特异性位点。标记好的探针(25 ng/mL)沸水浴10 min后立即冰浴10 min备用。倒掉预杂交液,加入50 ℃ 5 mL Hyb高效杂交液及变性地高辛标记探针(1~3 μL/膜,5~20 ng探针/mL Hyg杂交液)混匀,50 ℃杂交过夜。
取出尼龙膜50 ℃水浴震荡洗涤,室温封阻尼龙膜30 min、结合抗体30 min、洗抗体两次(15 min/次),加300μL NBT/BCIP显色底物于37 ℃培养箱中避光显色20 h后,短时间暴露于光下用50 mL ddH2O洗膜5 min,停止显色,拍照记录[8-12]。
2 结果
2.1 PCR法制备地高辛(DIG)标记探针
内生真菌sac探针电泳结果如图1所示,泳道1为非标记sac PCR反应产物,泳道2为DIG标记的sac探针,其电泳条带约709 bp,与预期结果相同。hph探针电泳结果如图2所示,泳道1为DIG标记的hph探针,泳道2为同样条件下的非标记PCR反应产物,电泳条带约879 bp,与预期结果相同。标记反应PCR产物中带非同位素的配体(生物素、地高辛等),其电泳迁移率慢于非标记PCR产物。
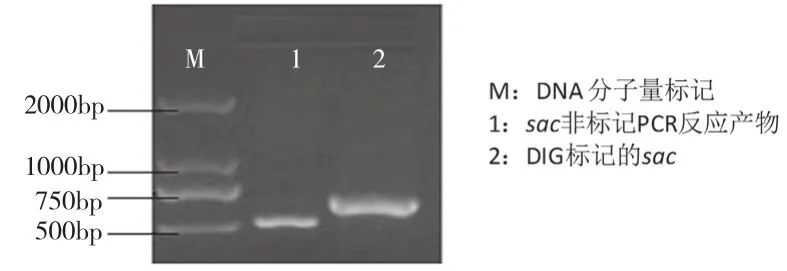
图1 OW7.8探针DNA琼脂糖凝胶电泳图
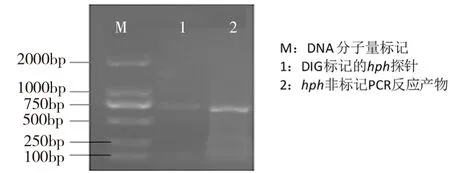
图2 M1探针DNA琼脂糖凝胶电泳图
2.2 OW7.8及M1基因组DNA限制性酶切与转膜
将提取的OW7.8和M1基因组DNA(终浓度为20.75±7.64 μg)用Eco RⅤ、Pst Ⅰ、Pci Ⅰ和Pvu Ⅱ限制酶进行切割,酶切产物浓度为202.6±13.8 ng/μL,OD 260/OD 280在1.88~2.01,电泳结果如图3所示。其中泳道1为野生株基因组DNA,泳道2、3为用限制酶Eco RⅤ、Pst Ⅰ酶切OW7.8基因组DNA后产物,由图可见酶切后泳道呈弥散状,无明显条带,说明酶切效果较好。泳道4、5为使用限制酶为Pci Ⅰ、Pvu Ⅱ酶切M1基因组DNA后的产物,结果显示泳道4酶切反应不完全,泳道5酶切反应完全。分别将OW7.8基因组用限制酶Eco RⅤ再进行酶切,将M1基因组用限制酶PciⅠ再进行酶切,结果如图4,可看到酶切反应进行彻底。转膜后观察凝胶结果如图5,凝胶上没有任何残留DNA,说明酶切产物已全部移至尼龙膜。

图3 基因组DNA酶切产物

图4 基因组DNA酶切产物

图5 转膜后的凝胶
2.3 Southern blot
由内生真菌sac特异性探针与M1基因组DNA和OW7.8基因组DNA酶切产物进行Southern blot试验,结果如图6所示。泳道1表示sac探针与Pvu Ⅱ切割M1基因组DNA杂交,杂交结果呈阴性,表明M1中sac已被敲除;泳道2和3分别用Eco RⅤ和Pst Ⅰ切割OW7.8基因组DNA进行杂交,杂交结果呈阳性,说明野生株有sac,且基因的拷贝数为1。
由hph特异性探针与M1基因组DNA和OW7.8基因组DNA酶切产物进行Southern blot试验,结果如图7所示。泳道1表示hph探针与Pvu Ⅱ切割M1基因组DNA杂交,发现M1杂交结果呈阳性,说明标记基因hph成功整合到M1基因组中;泳道2和泳道3分别用hph特异性探针与Eco RⅤ和Pst Ⅰ切割的OW7.8基因组DNA杂交,结果呈阴性,OW7.8基因组中无hph,M1中sac已被敲除,且标记基因hph已整合到基因组上。

图6 野生型探针杂交

图7 突变型探针杂交
3 讨论与结论
Southern blot过程较繁琐,需注意细节颇多,本研究用PCR法制备DIG标记探针时设计了6对特异性引物,经优化选择出两对特异性高且扩增产物长度适宜的引物,发现若扩增产物低于300 bp,则DIG掺入较少,而探针过长则DIG掺入过多,导致非特异性杂交。张明琴等[8]用900 bp探针进行杂交,杂交信号较强。另外,在探针标记前要优化PCR反应条件,包括引物、退火温度、延伸时间、模板量等,减少非特异性扩增,从而降低杂交背景值,避免出现假阳性。
Southern blot对基因组DNA的质量要求高,本研究在CTAB法基础上进行了改进[9],菌丝研磨要充分快速(2 min内),避免降解;涡旋振荡使菌丝与裂解液混匀,使DNA充分释放。对裂解时间也进行了探索,发现40 min利于基因组DNA释放。
实验中在45~65 ℃范围内设置了5个温度,发现50 ℃时杂交效果最好。杂交膜的选择也相当重要,试验发现使用尼龙膜比硝酸素纤维膜的杂交信号强。杂交液纯度也会影响杂交结果,分别用过滤前后的杂交液进行杂交,发现过滤后的杂交背景低、无假阳性、杂交信号较强,而未过滤杂交液则背景值高、无杂交信号。杂交膜用TE漂洗2次后封存,膜干燥或久置后,信号将会有所减弱或褪色,但经TE润湿后仍然会重现原有的杂交信号[10]。
Hyb高效杂交液适合在带正电荷尼龙膜上进行Southern和Northern杂交分析,其独特配方和操作规程仅需杂交6~12 h,而常规杂交则需要12~24 h。另外,Hyb高效杂交液能明显降低背景,使Southern膜上单拷贝基因和Northern膜上低拷贝RNA得以检出,也适于各类cDNA表达芯片杂交和探针标记。
设计DIG标记探针,利用Southern blot技术检测了小花棘豆内生真菌OW7.8和M1菌株,结果表明OW7.8中有sac中间序列,而在M1中被hph替换,说明sac已被敲除。sac基因以单拷贝形式存在于OW7.8基因组中。
猜你喜欢
装备制造技术(2020年1期)2020-12-25
数学大王·低年级(2020年8期)2020-08-14
湖北农机化(2020年4期)2020-07-24
世界农药(2019年4期)2019-12-30
今日农业(2019年11期)2019-08-15
生物工程学报(2019年1期)2019-01-30
乡村地理(2018年2期)2018-09-19
小雪花·初中高分作文(2016年9期)2016-05-14
中国与非洲(法文版)(2015年4期)2015-11-09
兵工学报(2012年8期)2012-02-23
