
Surface-confined Pt-based catalysts for strengthening oxygen reduction performance
2020-01-03 15:18:28YaoNieZidongWei
Yao Nie ,Zidong Wei
a Chongqing Key Laboratory of Inorganic Functional Materials,College of Chemistry,Chongqing Normal University,Chongqing,401331,China
b The State Key Laboratory of Power Transmission Equipment &System Security and New Technology,College of Chemistry and Chemical Engineering,Chongqing University,Chongqing,400044,China
ABSTRACT Developing highly efficient catalysts for the oxygen reduction reaction (ORR) is a key to the fabrication of commercially viable fuel cell devices for future energy applications.Considerable progress has been made to reduce Pt usage and improve performance of the Pt catalysts by modulating exposed facets of Pt nanocrystals and combining Pt with other metals to generate bimetallic nanocrystals with structures in the form of alloys,coreshells,branches or anisotropies.Apart from the above methods,confining Pt-based nanoparticles (NPs) surfaces with elaborately selected layers such as polymers,silicon or carbons can also lead to an optimized Pt electronic structure,which is beneficial to ORR process.In this minireview,we summarize the recent advancements in the area of surface-confined Pt-based electrocatalysts for ORR with emphasis on introducing the design strategies and synthesis methodologies.The integration of these catalysts into ORR operations and the resulting performance as well as the strengthening mechanisms is also discussed.Meanwhile,the insights into the research directions are proposed in order to shed light on the future development of surface-confined Pt-based ORR catalysts.
Keywords:Oxygen reduction reaction Fuel cell Surface-confined Pt-Based electrocatalysts
1.Introduction
Proton exchange membrane fuel cells(PEMFCs)have attracted much attention because of their high efficiency,high power density and zero emission of pollutants.Commercialization of PEMFCs has made tremendous progress and manufacturers such as Hyundai and Toyota have recently launched commercial fuel cell electric vehicles(FCEVs)on the market.However,it is still difficult to realize large scale applications of PEMFCs systems because the challenge of developing active but economical electrocatalysts to mitigate the sluggish kinetics of the cathodic oxygen reduction reaction (ORR).According to the report of the US Department of Energy (DOE),over 40% of the current cost of PEMFCs arises from Pt catalysts used for the ORR[1].On the other hand,satisfying PEMFCs lifetime requirements of 8000 h for cars and 20,000 h for buses,with<10%performance loss,still remains a grand challenge owing to catalyst deactivations[2].Generally,degradation of the most commonly used ORR electrocatalysts,namely,Pt-based NPs supported on carbons,predominantly happens via oxidation,dissolution,agglomeration of NPs and corrosion of carbon support[3-6].
Extensive research has been directed to developing efficient Pt-based catalysts with less precious metal loading and cost while preserving or achieving higher activity and durability during prolonged exposure to reaction conditions[7-10].As ORR is a heterogeneous catalysis process that is very sensitive to the surface properties of the catalysts,optimizing the surface electronic structure and atomic arrangement or coordination of Pt is believed to be effective to enable enhancement in both activity and durability.Accordingly,an altered electronic property would induce a change in adsorption behavior,especially making the formation potentials of surface-oxides shift positively and hence suppressing adsorption of oxygenated intermediates on Pt surface [11-13].This changed adsorption behavior is believed to be the origin of the high activity for ORR [14].In general,modulating surface electronic structures for Pt can be achieved by controlling exposed facets of Pt nanocrystals [15-17],alloying Pt with other metals [18-23],employing novel robust supports to induce a synergistic cocatalytic effect via a support-metal interaction[6,24-27].Among them,mixing Pt with other cheaper metals to form Pt alloy NPs has been most widely investigated as alloying can efficiently reduce Pt content and enhance catalyst performances through ligand and strain effects [19-21].
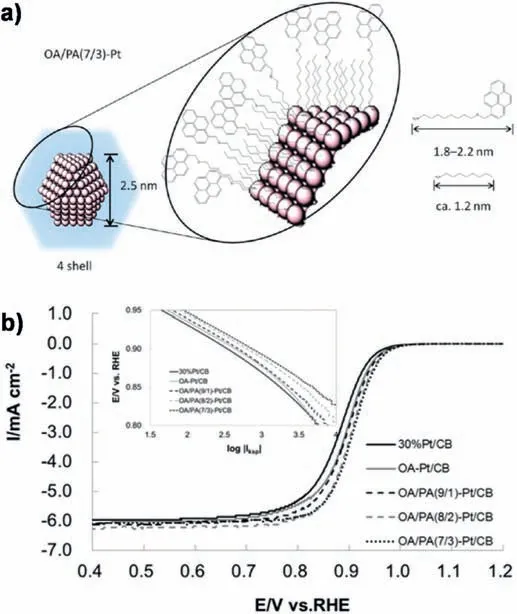
Fig.1.a) Schematic image of OA/PA (7/3)-Pt NP.One organic protective molecule occupies 1.32 surface Pt atoms.b)ORR polarization curves for OA-Pt/CB,OA/PA(9/1)-Pt/CB,OA/PA(8/2)-Pt/CB,OA/PA(7/3)-Pt/CB,and 30%Pt/CB catalysts recorded in an O2-saturated 0.1 M HClO4.Inset shows Tafel plots obtained from the polarization curves in the positive-going scans,which were normalized to the Pt electrochemical surface area.Reprinted with permission from Ref.[35].Copyright © 2014 American Chemical Society.
Apart from above routes,modifying Pt NPs surface with elaborately selected foreign species,such as organic polymers,silica or carbons can also lead to an optimized Pt surface electronic/geometric structure as well as favorable chemisorption properties.Despite the coverage of Pt catalytic sites,in some cases,this strategy can result in boosted ORR catalytic performances due to the unique interactions between coating layers and Pt NPs [7,28-30].Moreover,when such a surface confining strategy is applied into Pt-based alloy NPs,the activity and durability of inner Pt alloy would be further reinforced.This is of big significance as Pt alloy NPs are the most commonly used and intensively studied ORR catalysts at present.Additionally,preparation of some surface coating layers require high temperature heat-treatments (e.g.,carbon-based coatings),which may simultaneously arouse atom rearrangements within inner Pt NPs and thus result in a fascinating texturing and coordination states,leading to more strengthened ORR catalytic efficiency[31,32].
In this minireview,we summarize the recent advancements in the area of surface-confined Pt-based electrocatalysts for ORR with emphasis on introducing the design strategies and synthesis methodologies.The reinforcement mechanisms of coating layers as well as the integration of these catalysts into ORR operations and the resulting performance are also discussed.Meanwhile,the insights into the research directions are proposed in order to shed light on the future development of surface-confined Pt-based ORR catalysts.
2.Surface confined with organic compounds or polymers
In conventional opinion,the presence of organic ligands (capping agents) on NPs catalysts has been often regarded as a hindrance for applications in heterogeneous catalysis as they bind to the surface,leading to partial blocking of active sites.Thus the application of colloidal NPs for the preparation of model catalysts is most often preceded by removal of the ligands[33,34].In the last years,many studies have proved that organic ligands or compounds (e.g.alkylamines,aromatic compounds) attached to Pt NPs can block the adsorption of poisoning species,thus enhancing their activity for the ORR [35-41].For example,Miyabayashi and Miyake et al.[35]successfully modified Pt NPs surface with a controlled ratio of pyrene group (PA) and octylamine(OA)(9:1,8:2,and 7:3)by convenient two-phase liquid reduction procedures(Fig.1a).Such modification leaded to improvements in both ORR activity and durability compared to naked Pt NPs(Fig.1b),which might be contributed to a change of the adsorption kinetics of reaction intermediates.Zhou et al.[39]prepared chlorophenyl-functionalized Pt(Pt-ArCl)NPs(core dia.1.85±0.28 nm),and observed a 3 times higher electrocatalytic activity for ORR than that of bare Pt/C catalysts.The authors also suggested that the enhanced catalytic activity may be correlated with the impeded adsorption of oxygen species(e.g.,Oadand OHad)by chlorophenyl groups on Pt surfaces.Recently,Yamazaki et al.tried to clarify whether only a few planar macrocyclic compounds would modulate the ORR activity of Pt NPs [38].Interestingly,they demonstrated that a porphyrazine (2,7,12,17-tetra-(tert-butyl) tetraazaporphyrin,tBuTAP)can be adsorbed on a Pt NP surface via the N atoms in tBuTAP.Such an adsorbed tBuTAP molecule decreased the electrochemical surface area but increased the ORR mass activity of Pt NP by about two times.Cyclic voltammograms under argon atmosphere indicated that the onset potential for OHadgeneration shifts towards positive direction,implying that the tBuTAP impedes the adsorption of OHad on Pt NPs surface.
It can be clearly seen from above works that the adsorption behavior on Pt surfaces plays an important role in determining ORR activity.In fact,water molecules are considered as a major source of unfavorable oxygenated intermediates in catalysis of ORR [42-45].Jinnouchi et al.have proposed,based on their combined DFT and Poisson-Boltzmann calculations,that the interface water itself may also lead to blockage of Pt sites [46].The difficulty to improve ORR kinetics thus lies in the dilemma that water is the reaction media as well as the reaction product but might also cause blockage of active sites.Therefore,modifying the catalyst surface to be hydrophobic to perturb the H2O network may enhance the ORR activity.At this point,the hydrophobic ionic liquid(IL) may be a better choice than usual organic compounds,as the IL phase is supposed to partially replace water as the reaction media and more importantly,the created hydrophobic microenvironment at catalyst surfaces would help to preserve active sites by repelling water molecules from the product and aqueous electrolyte[47].Additionally,encapsulating Pt NPs surface with IL may increase O2solubility in IL-confined space,thus resulting in improved reaction kinetics[47,48].Several IL-modified Pt catalysts has already been constructed and exhibited enhanced activity and stability for ORR[47-51].For example,Erlebacher et al.reported the synthesis of a hydrophobic IL ([MTBD][beti])encapsulated nanoporous NiPt NPs catalyst[48](Fig.2a and b).The high O2solubility in the IL combined with the confined environment within the pores of NiPt NPs led to enhanced attempt frequencies and improved ORR kinetics.The half-cell specific activity of this catalyst was found to be an order of magnitude higher than that of commercial Pt/C(Table 1).Etzold et al.[47]used([C4C1im][NTf2])to modify the Pt/C surface and observed significantly reduced ORR overpotential and enhanced ORR activity,which would originate from the higher accessibility of Pt defect sites protected by the hydrophobicity conveyed by IL.Moreover,the authors found that the presence of the IL phase makes Pt surfaces more tolerant to poisonous species(OHad,Fig.2e and f)and its activity can be stabilized even after 30,000 aging cycles.
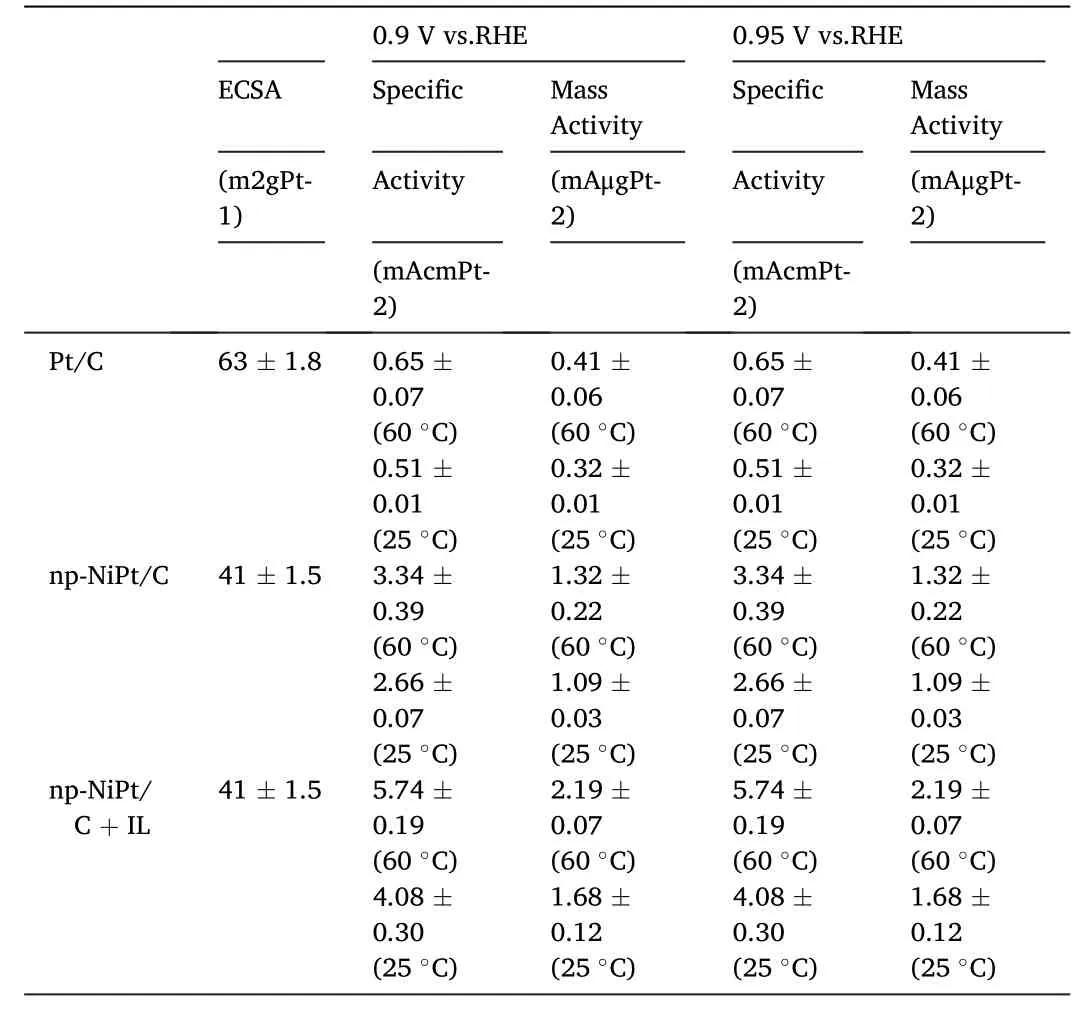
Table 1 Specific and mass activity numbers measured in the half cell at 60 °C and 25 °C for different catalysts mentioned in Fig.2a-c.Reprinted with permission from ref.[48].Copyright©2013 WILEY-VCH Verlag GmbH &Co.KGaA,Weinheim.

Fig.2.a)Schematic and implementation of[MTBD][beti]IL-encapulsated nanoporous NiPt.b)HRTEM of nanoporous NiPt NPs encapsulated with[MTBD][beti]IL supported on carbon.c) ORR curves for different catalysts recorded in O2 saturated 0.1 M HClO4 at 60 °C.Reprinted with permission from Ref.[48].Copyright ©2013 WILEY-VCH Verlag GmbH&Co.KGaA,Weinheim.Scheme for the interfacial microenvironments at d)Pt/C,and e)Pt/C-[C4C1im][NTf2],showing that the IL would selectively locate at the defect sites and protect the Pt sites from hydroxy species.Reprinted with permission from Ref.[47].Copyright © 2016 WILEY-VCH Verlag GmbH &Co.KGaA,Weinheim.
In order to modulate the chemisorption properties of Pt more significantly,some advanced polymers have been introduced onto Pt NPs surface.Typically,Wei et al.[29]synthesized a conducting polymer(polyaniline)coated Pt/C core-shell catalyst(Pt/C@PANI)via an in-situ polymerization method (Fig.3a) and its stability in both half-cell and single cell was significantly strengthened compared with uncoated Pt/C.Specifically,under solution test condition,the ECSA of Pt/C@PANI catalyst decreased by only~30% after 1500 CV cycles,whereas the naked Pt/C lost~83% of its initial ECSA (Fig.3b).While in the single cell,the cell current of PANI@Pt/C at 0.6 V decreased by only 1.3 times after 5000 cell cycles (Fig.3c),whereas that of Pt/C decreased by 7.1 times (Fig.3d).The authors attributed the enhancements to an electronic interaction between Pt and PANI,as demonstrated by X-ray photoelectron spectroscopy(XPS)analysis and DFT calculations.In XPS,the Pt 4f7/2 peak of Pt/C@PANI was shifted to a higher binding energy relative to that of the Pt/C,indicating an electron transfer from Pt to PANI (Fig.3e).This electronic interaction might alter the electronic structure of the Pt NPs,making it difficult for the Pt NPs to be oxidized.The DFT further disclosed that the system energy markedly decreased after covering PANI onto the Pt/C,meaning that Pt/C@PANI was more stable than Pt/C[30].The DFT calculations also demonstrated that the HOMO energy level in Pt/C@PANI lifted while the d-band lowered compared with Pt/C,thus reducing the gap between the HOMO of Pt/C@PANI and the LUMO of oxygen.This change favored for electron transfer between Pt/C@PANI and O2and facilitated desorption of intermediate species on Pt surface.Recently,Masafumi et al.[52]coated poly (melamineco-formaldehyde) methylated onto Pt/C catalyst and a positively shift in redox peaks of the OHadadsorption/desorption associated with an increased ORR activity were observed.
3.Surface confined with silicon-based layers

Fig.3.a)Configuration of the Pt/C@PANI Catalyst.b)Pt 4f XPS spectra of the Pt/C and Pt/C@PANI catalysts.Reprinted with permission from ref.[29].Copyright©2012,American Chemical Society.Reprinted with permission from Ref.[29].Copyright © 2012 American Chemical Society.

Fig.4.a) b)TEM images of SiO2/Pt/CNT (MTEOS).c) Polarization curves for the ORR on fresh Pt/CNT,SiO2/Pt/CNT (TEOS) and SiO2/Pt/CNT (MTEOS).Polarization curves for the PEMFC single cells with the d) Pt/CNT,e) SiO2/Pt/CNT (TEOS) and f) SiO2/Pt/CNT (MTEOS) cathode catalysts during the durability tests.Reprinted with permission from Ref.[58].Copyright © 2014 American Chemical Society.
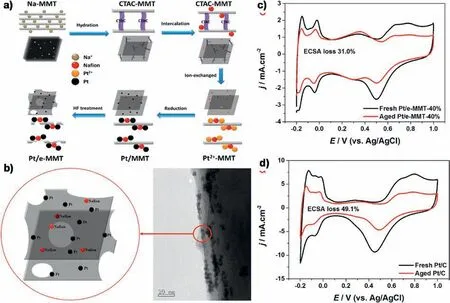
Fig.5.a)Schematic representation of the synthesis of Pt/e-MMT.b)Structure scheme and TEM image of Pt/eMMT.Cyclic voltammograms for c)Pt/e-MMT and d)Pt/C before and after 1600 cycles in N2-saturated 0.5 M H2SO4 solution.Reprinted with permission from Ref.[60].Copyright © 2016 Science China Press and Springer-Verlag Berlin Heidelberg.
A series of works about SiO2-covered Pt catalysts was conducted by Takenaka’ group [53-59].As early as in 2007,Takenaka and his co-workers [57]have studied the catalytic performance of multi-walled carbon nanotube (CNT)-supported Pt (Pt/CNT) cathode catalysts covered with silica layers and found that the silica-coated Pt/CNT(SiO2/Pt/CNT) exhibited high durability under the PEFC cathode conditions,whereas Pt/CNT without a silica-coating became significantly deactivated under the same conditions.The durability enhancement was mainly attributed to the protective effect of the silica coverage,as it significantly prevented the migration of Pt NPs and the diffusion of dissolved Pt species out of the silica layers.Despite the improved stability,the ORR catalytic activity of SiO2/Pt/CNT for ORR was slightly inferior to that of Pt/CNT.This was because the silica coverages act as diffusion barriers that hinder the mass transport of reactants and products.Aiming at addressing this issue,Takenaka et al.tried to make the silica coverage hydrophobic and porous to promote diffusion of oxygen and water molecules.They experimentally realized this idea by successive hydrolysis of 3-aminopropyltriethoxysilane (APTES) and methyltriethoxysilane (MTEOS),rather than previously used tetraethoxysilane (TEOS),in presence of Pt/CNT [58].The as-formed silica coverage was very thin (2-3 nm) (Fig.4a and b) and29Si-NMR spectra analysis directly demonstrated that it indeed has hydrophobic methyl groups,which would enhance the discharge of water molecules outside of the silica layers.Additionally,SiO2/Pt/CNT catalysts prepared from APTES and MTEOS owned porous structures with larger pore diameters than those prepared from APTES and TEOS,which favors for accelerating the diffusion of molecular oxygen to the inner Pt surfaces.As expected,the SiO2/Pt/CNT (MTEOS) showed not only superior stability but also higher ORR activity than the SiO2/Pt/CNT(TEOS)under half cell tests (Fig.4c).The catalytic activity and durability of SiO2/Pt/CNT catalysts were also evaluated in PEMFC single cells.As shown in Fig.4d-f,both the fresh SiO2/Pt/CNT showed lower power output than fresh Pt/CNT.However,the catalytic activity of these SiO2/Pt/CNT catalysts was gradually improved with the cycle number up to 1000 cycles,whereas the Pt/CNT was seriously deactivated,further confirming the protection and stabilization effect of SiO2.As for the two different SiO2/Pt/CNT catalysts,the SiO2/Pt/CNT (MTEOS)exhibited a higher cell current than SiO2/Pt/CNT(TEOS)in the region of lower cell voltage,which can be owing to the porosity of SiO2endowed by the MTEOS.
Furthermore,Takenaka et al.also tried to add pore-making templates,namely NH2-(CH2)n-NH2(n=6,8 and 10),during the preparation of SiO2-covered Pt/CNT catalysts to further engineer the pore structure in silica layers,and attained positive experimental results[59].Although SiO2shells have shown to prolong the catalyst lifetime,they only seem like physical barriers and whether they intrinsically modify the electronic structure of Pt surface or not have been not investigated deeply.
Besides SiO2,a natural silicate,montmorillonite (MMT) has been also applied to enhance the stability of Pt NPs [60].As illustrated in Fig.5a,a sodium modified montmorillonite (Na-MMT) was firstly ion-exchanged with cetyltrimethylammonium chloride (CTAC) to obtain CTAC modified MMT (CTAC-MMT) and then was intercalated with cetyltrimethylammonium ions (CTA+) to enlarge the layer distance of CTAC-MMT.The attained CTAC-MMT was further mixed with Nafion to improve the proton conductivity.After the ion exchange of Ptwith CTA+,an in situ chemical reduction reaction of Pt was carried out inside the layers and thus Pt NPs were immobilized between the MMT layers(Pt/MMT).Finally,the Pt/MMT was partial etched in a 40% HF solution to obtain Pt/e-MMT (Fig.5b).Due to the space-confinement effect of MMT,the as-prepared Pt/e-MMT catalyst exhibited significantly improved stability than commercial Pt/C catalyst.As directly evidenced in Fig.5c and d,the Pt/e-MMT showed 31.0%diminution in the Pt ECSA,whereas the Pt/C catalyst loses approximately 49.1% of its initial ECSA after 1600 cycling.The X-ray photoelectron spectroscopy (XPS) analysis found that the Pt 4f peaks in Pt/e-MMT shift to higher binding energy by 0.54 eV compared with those of commercial Pt/C,indicating the interaction between Pt NPs and e-MMT is promoted.However,the exact interaction and the definite promotion mechanism were not illustrated clearly in this work,which need other means such as DFT calculation to verify.

Fig.6.a)Synthetic Process of Pt/C@NGC.HRTEM images of b)Pt/C@PANI and c)Pt/C@NGC.Reprinted with permission from Ref.[61].Copyright©2014 Royal Society of Chemistry.d)Synthetic Process of PtNi/C@NC.e)HRTEM images of PtNi/C@NC.Reprinted with permission from Ref.[62].Copyright©2019 Hydrogen Energy Publications LLC.f) Synthetic Process of Pt/CNTs@C.g) HRTEM images of Pt/CNTs@C.Reprinted with permission from Ref.[63].Copyright © 2017 American Chemical Society.i)-l) HRTEM images of Pt@CNx/CNT.m) The statistical pie diagram of Pt nanocrystals in different status in Pt@CNx/CNT catalyst.Reprinted with permission from Ref.[28].Copyright © 2015 American Chemical Society.
4.Surface confined with carbon layers
Another widely used protecting coating for Pt NPs is carbon layers.In general,the carbon shells are formed by carbonization of specific polymers at appropriate temperature under inert atmosphere.For example,we have employed polyaniline (PANI) as the precursor and polymerized them in-situ on the surface of the Pt/C catalyst,followed by subjecting Pt/C@PANI to carbonization under N2atmosphere at 900°C to prepare a nitrogen-doped graphitic carbon(NGC)coated Pt/C catalyst(Pt/C@NGC,Fig.6a) [61].The high-resolution transmission electron microscopy (HRTEM) images clearly demonstrate the core-shell structure of the Pt/C@NGC catalyst before and after heat treatment,as shown in Fig.6b and c.It can be clearly seen that the thickness of decoration layer for Pt/C@PANI and Pt/C@NGC shrinks from 11 nm to 3 nm,which was owing to the substantial PANI decomposition during heat-treatment.It is worthy to point out that the confinement effect of the PANI could render Pt NPs with an anti-sinter capability and the size of Pt NPs in Pt/C@NGC catalyst increased only~2 nm even after 900°C heat treatment,thus guaranteeing the feasibility of the design concept of the Pt/C@NGC.By virtue of the NGC protection,the as-constructed Pt/C@NGC catalyst exhibited evidently enhanced stability when compared with the uncoated Pt/C catalyst,as the former showed 8%diminution in the Pt ECSA and only a 16 mV degradation in the half-wave potential,whereas the loss of Pt ECSA is as high as 41% and the shift in the half-wave potential was 61 mV for the later.Despite the coverage,the NGC would not block the transport of the aqueous electrolyte to the inner Pt active sites due to its porous nature.Thus the Pt/C@NGC catalyst also exhibited an enhanced ORR activity with a specific activity of 0.308 mAcm-2and a mass activity of 163 mAmg-1Ptat 0.9 V,which was 2.5 times and 1.7 times greater than those of Pt/C respectively.
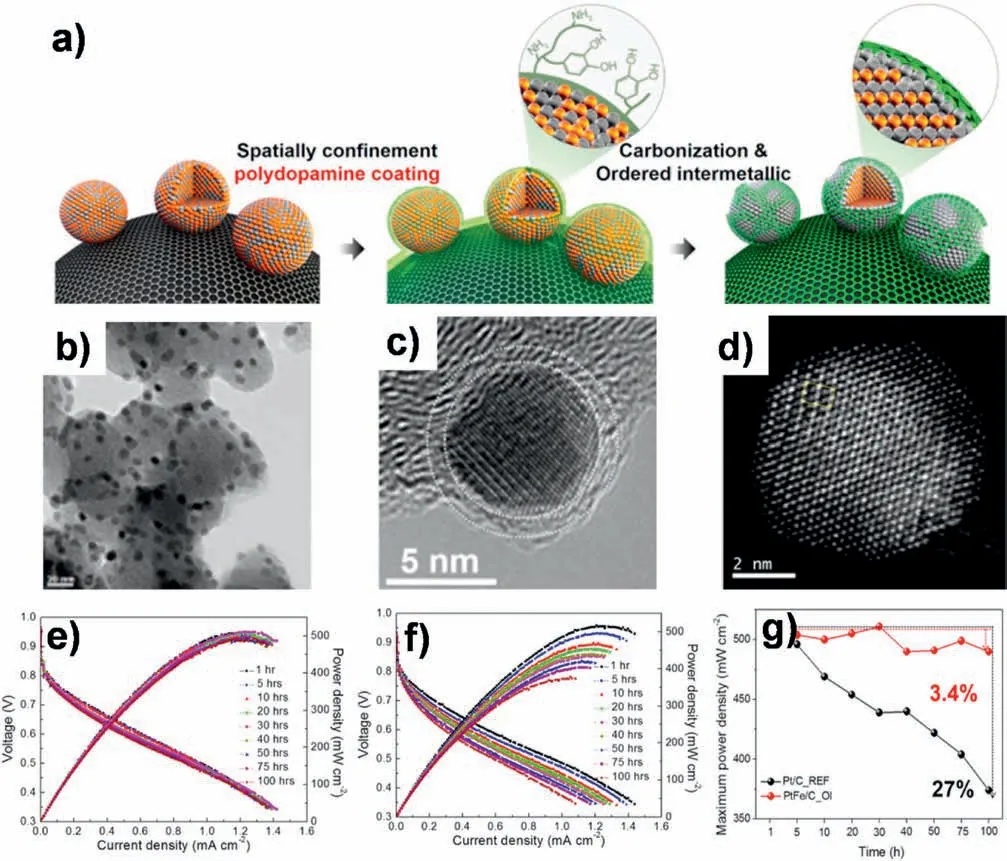
Fig.7.a) Synthesis of NC-coated ordered fct-PtFe.b)TEM and c)HRTEM images of NC-coated ordered fct-PtFe.c)Cs-corrected HAADF-STEM image of NC-coated ordered fct-PtFe.Results from 100 h MEA test for (e) NC-coated ordered fct-PtFe and (f) Pt/C.g) Maximum power density plot of 100 h MEA test as functions of operating time for NC-coated ordered fct-PtFe and Pt/C.Reprinted with permission from Ref.[31].Copyright © 2015 American Chemical Society.
Since Pt alloy NPs supported on carbon are the most commonly used ORR catalysts at present,improving the stability and activity of them is urgently desirable.Thus,we have further applied above in-situ PANI polymerization-carbonization method to promote the stability and activity of solution phase synthesized carbon-supported PtNi NPs(Fig.6d)[62].As the same as the case of preparing Pt/C@NGC,the in situ polymerized PANI shell also made the PtNi NPs highly resistive toward sintering and the average size of resultant PtNi NPs in the synthesized PtNi/C@NC increased only~3 nm compared to that of initial PtNi/C.As expected,the as-synthesizedN-doped carbon confined PtNi/C catalyst(PtNi/C@NC,Fig.6e) exhibited much enhanced ORR stability and activity compared to initial PtNi/C and commercial JM-Pt/C catalysts.We found the binding energy of Pt 4f peaks in high-resolution XPS signals for PtNi/C@NC shifted positively by~0.71eV relative to that for PtNi/C,implying there was an electronic interaction between NGC and Pt NPs.This interaction would result in a weaker adsorption of intermediate oxide species and thereby creating a stabilizing effect against Pt oxidation/dissolution,which may be the origin of the enhanced catalytic behavior.Such an electronic interaction between Pt NPs and carbon coating layers was also demonstrated by Guo et al.‘s work [28].They successfully embedded Pt NPs intoN-doped porous carbon/carbon nanotubes(Pt@CNx/CNT,Fig.6i-l)via a facile in situ reduction process and significantly improved ORR stability and activity of Pt@CNx/CNT compared to those of commercial Pt/C were observed.
It can be easily inferred that the thickness of carbon-coating layers play an important role in influencing the ORR catalytic behavior of final covered Pt-based catalysts.Specifically,un-continuous or too thin carbon-coating layers would fail in effectively protecting the Pt NPs,while too thick ones lead to the blockage of the inner Pt catalytic sites.Experimental results indicated that when the thickness of the NC shell was below 1 nm,an excellent protection of the NPs as well as high catalytic activity can be achieved.In general,the thickness of the carbon coating can be controlled by varying the amount of added polymers.Sun et al.prepared carbon-encapsulated Pt/CNTs(Pt/CNTs@C)catalysts by a facile in situ polymeric encapsulation-graphitization method and conducted a precisely control over carbon thickness [63] (Fig.6f).By simply programming the polymer growth on Pt/CNTs,the thickness of the carbon-coating layer can be tuned from 0.5 nm to several nanometers.Electrochemical evaluations indicated that the electrocatalytic activity strongly depends on the thickness of the carbon-coating layers,and the Pt/CNTs@C with an ultrathin carbon layer thickness of~0.8 nm(corresponding to~2-3 graphene layers,Fig.6g)showed the best ORR activity,and excellent durability with no noticeable activity loss even after 20 000 cycles of accelerated durability tests.Very recently,Karuppannan et al.[64] encapsulated Pt NPs supported on carbon nanofibers (CNFs) into a subnanometer scaleN-containing carbon layer by using a Pt-aniline complex coated CNFs precursors.Heat-treating the coated CNFs produced 3-4 nm-sized Pt NPs that were uniformly coated with a~1 nm carbon layer on the CNF surface (Pt@CS/CNF).The stability and catalytic activity of this Pt@CS/CNF for ORR are high owing to the robustness of the carbon shells that secure the Pt NPs.In a unit cell test,the performance of Pt@CS/CNF heat-treated at 900°C was almost maintained for 30,000 accelerated stability test cycles,showing a negligible voltage loss at an operating current density of 0.8 A cm-2.

Fig.8.Schematic fabrication of H-PtFe/C@NC based on space-confned pyrolysis and nanoscale Kirkendall effect.Reprinted with permission from Ref.[65].Copyright © 2016 WILEY-VCH Verlag GmbH &Co.KGaA,Weinheim.
Despite the achievements in controlling the carbon coverage thickness,highly variable results still exist in preparing carbon-coated Ptbased catalysts.An important information in Guo et al.‘s work should be mentioned herein that according to the statistical analysis of over 200 Pt NPs at the edges of CNT in Pt@CNx/CNT,71.23% of the Pt NPs were fully coated with continuous carbon layers (Figure 6m),while the rest were coated with a few fractured carbon layers,meaning that coating all NPs with a uniform and continues carbon layers is very difficult.More precise and advance synthesis protocols are therefore required in the future.
Since yielding carbon coating layers needs high temperature heattreatments,the inner Pt-based NPs also have to undergo a confined annealing process,which may lead to atom rearrangements within NPs and thus result in a more ordered and stable phase in NPs.Chung et al.prepared highly durable and active PtFe NPs with a very thin“dual purpose”N-doped carbon (NC) shell [31].This NC shell was in situ formed from a polydopamine coated on disordered fcc-PtFe NPs and these disordered NPs could be transformed into an ordered fct structure during the annealing process (Fig.7a).The so-called“dual purpose”means that the NC shell not only prevented the coalescence of the NPs during a thermal annealing to keep the size of resultant ordered fct-PtFe NPs as small as 6.5 nm (Fig.7b and c),but also protected them under harsh fuel cell operating conditions.When integrated into a membrane electrode assembly (MEA) and performed continuous operation of 100 h,the fct-PtFe/C coated with an NC shell clearly demonstrated the higher stability with the loss in the maximum power density of only 3.4%,while the loss for commercial Pt/C catalyst was 27%(Fig.7e-g).In situ XANES,EDS,and first principles calculation studies disclosed that both an ordered fct-PtFe structure and the NC shell are critical for the excellent performance.Specifically,the Fe atoms in ordered fct-PtFe structure were more stable and less dissolved than those in disordered fcc-PtFe,and the NC shell could stabilize Pt-rich surface of the NPs due to strong adsorption of nitrogen to Pt atoms.
Structure evolution of NPs from solid into hollow can be also realized within carbon coverages.We have directly transformed solid Pt NPs into hollow PtFe alloys featuring with a NC shell coating through spaceconfned pyrolysis and nanoscale Kirkendall effect [65].In this synthesis,the NC shell was derived from in-situ formed PDA layer and besides that,an removable silica layer was also employed.As illustrated in Fig.8a,the inner PDA layer was acted as a strong chelating agent to homogeneously adsorb Fe3+precursor,and the outer silica layer was applied to create a confined space for preventing NPs sintering and hindering PDA loss during high-temperature pyrolysis.Within this double-layers confined space,a nanoscale Kirkendall effect occurred under H2-assisted high-temperature pyrolysis.As the outward diffusion rate of Pt was faster than the inward diffusion rate of Fe,Pt atoms tended to enrich at surface accompanying with the formation of voids and hollows(Fig.8b).After etching the silica,hollow PtFe NPs with a Pt-skin texture covering by a NC shell were attained (Fig.9 a-f).The mass activity and specific activity of as-prepared H-PtFe/C@NC at 0.9 V reached 1.35 mA cm-2and 0.993 A mg-1Pt,respectively,which were much higher than those of the benchmark commercial Pt catalyst(0.268 mA cm-2and 0.193 A mg-1Pt).Similar to above works,with the protection of NC shell,H-PtFe/C@NC was electrochemically and geometrically stable,as it exhibited only 7 mV negative shift for its half-wave potential after 20,000 CV cycles (Fig.9g),and no noticeable NPs ripening or aggregation was observed and the hollow shape of PtFe NPs largely maintained after aging (Fig.9h).
Moreover,we further found that by tuning the feeding Pt/Fe ratio or the annealing temperature in above PDA-SiO2co-confined annealing synthesis,two kinds of NC covered PtFe NPs can be formed,i.e.,Pt-skinlike hollow alloy and ordered PtFe intermediates [66],as shown in Fig.10.This structure evolution within NPs was governed by the competition between the segregation energy and chemical ordering energy during high temperature.Similarly,by virtue of the inner PDA shell and the outer SiO2layer,the structure evolution can proceed smoothly without occurrence of severe NPs sintering.Electrochemical activity results revealed that when compared with the hollow disordered PtFe alloy and commercial Pt/C catalyst,the ordered PtFe intermediates exhibited the best ORR performance,with a specific activity of 2.03 mA cm-2and a mass activity of 1.48 AStability tests showed that after 30,000 CV cycles,the baseline Pt/C catalysts suffered from a substantial loss in specific surface area caused by NP ripening and aggregation,while in sharp contrast,no noticeable ECSA loss and NP aggregation were observed for eitherNC-coated ordered PtFe or disordered one.This result suggested that NC protecting shell formed in-situ makes a significant contribution in enhancing the electrochemically and geometrically stability.
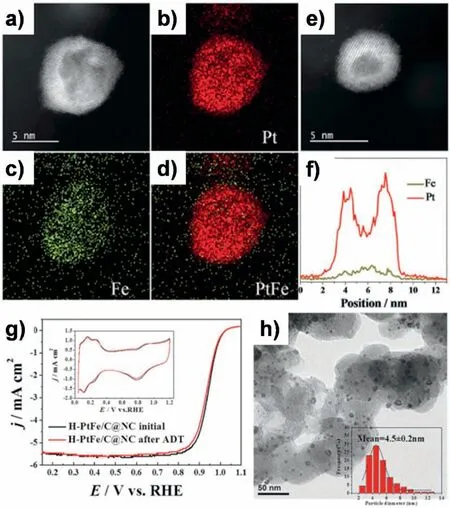
Fig.9.a)-d)HAADF-STEM image of a hollow PtFe NP and the corresponding STEM-EDX elemental mapping images of Pt,Fe,and PtFe.e)HAADF-STEM image of a hollow PtFe NPs and f)corresponding EDX line-scanning profile.g)ORR polarization curves of H-PtFe/C@NC before and after 20,000 potential cycles between 0.6 and 1.2 V versus RHE.h)TEM image and particle-size distribution of aged H-PtFe/C@NC catalyst after 20,000 CV cycles.Reprinted with permission from Ref.[65].Copyright © 2016 WILEY-VCH Verlag GmbH &Co.KGaA,Weinheim.
Those three works are of universality and can be readily applied to other Pt bimetallic or multimetallic systems,thus provides a new and versatile path to fabricating advanced and stable Pt-based NPs.
5.Conclusions
In summary,surface-confined Pt-based NPs with elaborately selected organic or inorganic matters emerged as a new class of highperformance electrocatalysts towards ORR.Owing to the interfacial interaction,this method may lead to a systematically tuned Pt surface electronic structure associated with an optimized adsorption behavior,enabling enhancement in both activity and durability.Specifically,the presence of organic ligands/molecules/compounds on Pt surface can result in an improved selectivity and ORR performance by affecting the adsorption of intermediates as well as reactant species.Such a positive effect may be induced by the steric,electrostatic,or hydrophilic/hydrophobic properties of external modified organic matters.However,these roles remain unclear and still needs to be probed.
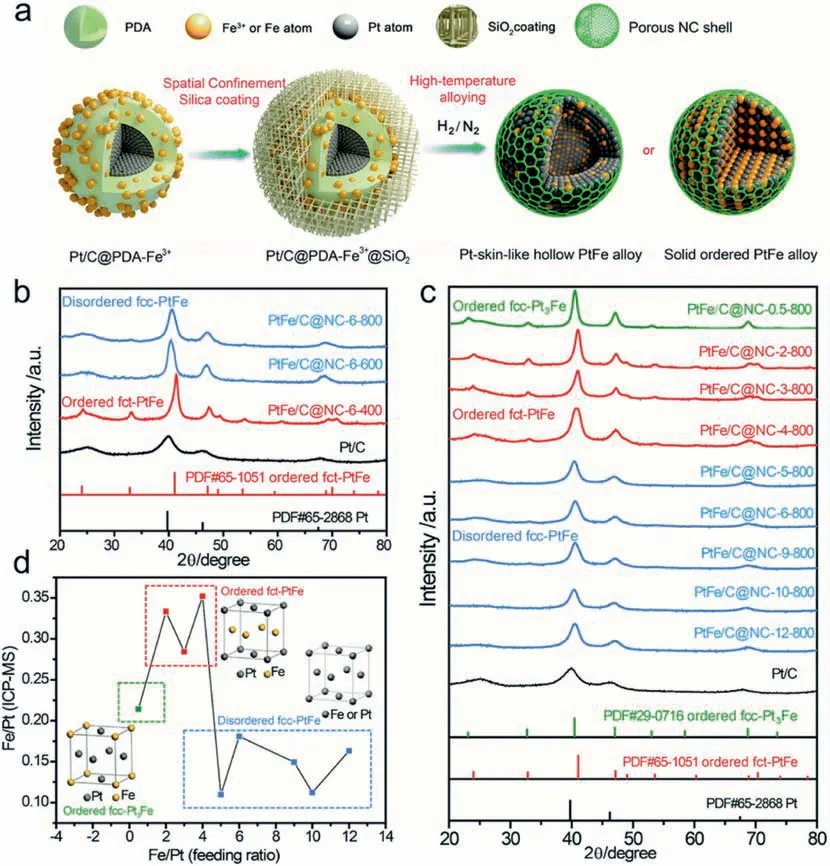
Fig.10.(a) Schematic illustration of the formation of two kinds of NC covered PtFe NPs.(b) XRD of PtFe alloy samples obtained with different annealing temperatures at a fixed feeding molar ratio of Fe3+ to Pt.For“PtFe/C@NC-X-T′′,where“X”represents the feeding molar ratio of Fe3+ to Pt and“T”represents the annealing temperature.(c) Powder X-ray diffraction patterns of PtFe alloy samples obtained with different feeding molar ratios of Fe3+ to Pt at 800 °C.(d) Relationship between the feeding molar ratio of Fe3+ to Pt and the corresponding molar ratio of Fe to Pt,as determined by ICP-MS.Reprinted with permission from Ref.[66].Copyright © 2019 Royal Society of Chemistry.
The improvement in stability via coating SiO2shells is apparent,however,it seems that SiO2shells just act as physical barriers and whether they intrinsically modify the electronic structure of Pt surface or not have not yet been investigated deeply.While many theoretical calculation results have manifested that the coating of some organic polymers and carbons will induce an electronic interaction between inner Pt-based NPs and outer shells.These unique interactions often alter the electronic structure of the Pt,making it difficult for Pt to be oxidized under ORR working conditions.Nonetheless,many those polymers and undoped carbons are inert,and thereby the coverage of them would decrease the catalytic activity.With respect to this,doped carbon protective layer,especially theN-doped carbon shows much superiority and has been extensively investigated.As the N atom incorporated promotes the activity of carbon matrix towards the ORR,confining Pt NPs inN-doped carbon not only reduces the ECSA loss of Pt during aging tests,but also achieves higher activity than that of bare Pt.The improved performance originates from the electronic interactions between nitrogen and Pt[67].Such an interaction enhances the binding energy between Pt and NC shell,and thus stabilizing Pt more efficiently.
Besides,the formed carbon coating layers are often endowed with porous nature due to the evaporation of small molecule fragments of precursors during carbonization.Moreover,the preparation of carbon coating layers requires high temperature carbonization treatments,during which fascinating atom rearrangements in metal NPs within nanoscale confined space may occur,and thus ensure the evolution of extremely stable and effective Pt-based NPs.
Despite the superiorities,the precisely control over the thickness of surface coatings is still a big concern as a layer that is too thick would hinder the access of reactants during electrocatalysis,while a too thin one cannot sufficiently protect the Pt.Although some ultrathin but robust coating layers has been achieved as reviewed in above,how to balance this two factors still remains a challenge.At this point,we should persist in seeking for advanced in-situ synthesis methods for preparing controllable surface coatings layers on Pt surface.Searching or designing coating layer precursors with easy-processivity,welldefined structure,suitable hydrophilicity/hydrophobicity is also desirable.Additionally,pore-making agents or templates can be used to achieve highly permeable of coatings layers,but it should be taken into consideration that adding and removing those agents and templates may complicate synthesis process and contaminate Pt surface.On the other hand,the development of theoretical calculations with proper structure models that close to the reality of surface-confined Pt NPs would be helpful in predicting the influence of the layers of surface coatings on ORR behavior,and thus providing the important clues for directing experimental fabrication.
Nowadays the reported number of surface-confined Pt catalysts is relatively less than other popular kinds of Pt-based ORR catalysts such as Pt alloys and polyhedrons,it is expected that more effective and economical Pt-based catalysts with surface modifications will be continuously proposed in the near future.In this regard,the present review imparts the positive prospect for future works on the elaboration of this type of ORR catalysts.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially sponsored by the International cooperation program of National Natural Science Foundation of China (Grant No.21761162015),the National Natural Science Foundation of China(Grant No.21802013),and the Top-notch youth Talent project of Chongqing Normal University(Grant No.02030307-0075).
 Progress in Natural Science:Materials International2020年6期
Progress in Natural Science:Materials International2020年6期
- Progress in Natural Science:Materials International的其它文章
- Key materials and technologies for fuel cells
- Experimental measurement of proton conductivity and electronic conductivity of membrane electrode assembly for proton exchange membrane fuel cells
- Nickel-introduced structurally ordered PtCuNi/C as high performance electrocatalyst for oxygen reduction reaction
- Large specific surface area S-doped Fe-N-C electrocatalysts derived from Metal-Organic frameworks for oxygen reduction reaction
- Surface modifications of Pt-based atomically ordered nanoparticles to improve catalytic performances for oxygen reduction reaction
- Accelerated Test of Silicone Rubbers Exposing to PEMFC environment