
儿童头颈部非横纹肌肉瘤软组织肉瘤临床研究
2019-12-23 08:21:18王生才张大伟王希思何乐建张建国马晓莉
首都医科大学学报 2019年6期
段 超 王生才 金 眉 张大伟 赵 文 王希思 赵 倩 邰 隽 张 杰 何乐建 张建国 倪 鑫 马晓莉*
(1. 国家儿童医学中心 首都医科大学附属北京儿童医院血液肿瘤中心 儿科学国家重点学科 儿科重大疾病研究教育部重点实验室,北京 100045; 2. 国家儿童医学中心 首都医科大学附属北京儿童医院耳鼻咽喉头颈外科,北京 100045; 3. 国家儿童医学中心,首都医科大学附属北京儿童医院病理科,北京 100045;4. 北京大学口腔医院颌面外科,北京 100081)
非横纹肌肉瘤软组织肉瘤(non-rhabdomyosarcoma soft tissue sarcoma, NRSTS)是一组罕见的恶性肿瘤,起源于间叶组织,美国癌症检测网评估,20岁以下青少年软组织肉瘤每年发病率为11.0人/100万[1],我国暂无统计资料。原发部位以四肢较多见,躯干及头颈部少见,多常见于青春期及青年。虽然婴儿期也可见NRSTS,但是在组织学上有其特殊的类型,如婴儿型纤维肉瘤等。头颈部解剖结构复杂,肿瘤位置通常毗邻重要器官,诊疗措施及原则较原发于其他部位的软组织肉瘤不同。儿童头颈部NRSTS约占儿童NRSTS的7%~18%[2],对此类疾病缺少系统的研究。
对成人的研究[2-5]显示,发生在头颈部的NRSTS局部复发率高,预后较发生于四肢的肿瘤差。但儿童NRSTS与成人相比,在组织病理类型及临床表现方面存在不同,但是目前国内尚无此方面的报道。我国目前对于非横纹肌肉瘤软组织肉瘤的诊治,包括病理分级、临床分期分组并无统一的规范,缺少大宗病例临床研究。本研究回顾性分析首都医科大学附属北京儿童医院单中心头颈部非横纹肌肉瘤软组织肉瘤的临床特点及治疗情况,提高对这一少见肿瘤的认识,为进一步规范非横纹肌肉瘤软组织肉瘤的诊治提供临床数据支持。
1 对象与方法
1.1 研究对象
采用病例观察的研究方法。本研究经首都医科大学附属北京儿童医院医学伦理委员会批准,批准文号:2018-K-106,所有入组患儿家长在治疗开始之前均签署知情同意书。所有入组患儿年龄均<18周岁。入组时间为2012年10月至2018年2月。
1)入组标准:新诊断的,未经过放射治疗(以下简称放疗)、化学药物治疗(以下简称化疗)的患儿;经病理诊断证实属于非横纹肌肉瘤软组织肉瘤;所有患儿在治疗前均经过首都医科大学附属北京儿童医院,以及北京大学第三医院两家三级甲等医院病理会诊;依据不同的病理类型针对性行相应的融合基因检测。
2)剔除标准:曾于外院接受过放化疗的患儿;治疗不足2个疗程非疾病进展放弃治疗者;病理诊断不明确者。
3)治疗前评估检查:入组患儿治疗前需行完善的影像学检查评估原发瘤灶及常见转移部位,包括:原发瘤灶部位的增强磁共振(magnetic resonance imaging,MRI)或增强计算机断层扫描(computed tomography,CT),胸部CT,头颅MRI,正电子发射断层显像(positron emission tomography, PET)/CT及骨扫描。病理为高度恶性患儿需行骨髓常规除外骨髓侵犯;瘤灶位于脑膜旁区或者脊柱旁区者,进行脑脊液检查。治疗期脏器评估:包括血、尿、便常规,血生化、心电图及心脏彩超、听力检查(应用铂类前)等。
4)患儿的临床特征,包括:性别、诊断年龄、病理类型、肿瘤大小、淋巴结转移情况、远处转移情况、治疗措施及随诊等资料的收集均来自患儿的病例资料及影像学检查结果。肿瘤特征的评估包括原发部位、肿瘤直径(≤5 cm,>5 cm)、病理亚型、组织病理级别、局部侵犯、淋巴结侵犯以及有无远处转移。
1.2 治疗方案
患儿均采用多学科联合诊治模式,包括手术、化疗及局部放疗。可以手术切除者,可先行手术治疗;不能手术切除者,先行化疗,之后再酌情手术切除或放疗。化疗方案依据不同病理类型,滑膜肉瘤、恶性外周神经鞘瘤及伴有CIC-DUX4易位不能分类的小细胞肉瘤均采用长春新碱+阿霉素+环磷酰胺(Vincristine+Doxorubicin+Cyclophosphamide, VDC)/异环磷酰胺+依托泊苷(Ifosfamide+Etoposide, IE)方案交替,婴儿型纤维肉瘤采用长春新碱+防线菌素D+环磷酰胺(Vincristine+Actinomycin D+Cyclophosphamide, VAC)/IE方案,孤立型纤维肉瘤采用VAC/异环磷+阿霉素(Ifosfamide+Doxorubicin, ID),恶性横纹肌样瘤参照美国儿童肿瘤协作组(Children’s Oncology Group, COG)高度恶性肾脏肿瘤UH-1方案进行,即长春新碱+环磷酰胺+阿霉素(Vincristine+Cyclophosphamide3+Doxorubicin, VDCPM3)/环磷酰胺+卡铂+依托泊苷(Cyclophosphamide5+Carboplatin+Etoposide, CPM5+CE)方案交替。局部放射治疗方式的选择包括外放疗及125I粒子植入治疗。
1.3 随诊管理
所有患儿治疗期间每2个疗程后行原发瘤灶部位的评估,每4个疗程需加做常见转移部位的评估。患儿治疗结束后每3~6个月于本院肿瘤内科专业门诊随诊,评估原发瘤灶、胸部CT及头颅MRI等检查。如发现有复发或转移,记录发生的时间及部位。
1.4 统计学方法
对符合入组标准的连续病例进行统计分析。计数数据采用频数和百分数描述。采用Kaplan Meier方法进行生存分析。
2 结果
2.1 一般资料
本中心收治头颈部非横纹肌肉瘤软组织肉瘤患儿11例,年龄1.1~12岁,中位年龄5.6岁,其中男性7例,女性4例;病理类型包括:恶性横纹肌样瘤3例,滑膜肉瘤2例,婴儿型纤维肉瘤2例,孤立型纤维肉瘤1例,恶性神经鞘瘤1例;伴有CIC-DUX4易位的小圆细胞肉瘤1例,未分类的小圆细胞肉瘤1例。2例婴儿型纤维肉瘤患儿行ETV6-NTRK3融合基因检测,结果均为阴性。2例滑膜肉瘤患儿SYT-SSX融合基因均为阳性。原发部位:颈部4例,头皮及面部软组织3例,口腔、上颚、口咽3例,颞下窝1例;肿瘤直径≥5 cm为6例,<5 cm为5例;6例患儿无区域淋巴结转移,5例存在区域淋巴结转移;有远处转移者3例,余8例患儿不存在远处转移,其中2例患儿为肺转移,1例患儿为骨、骨髓、胰腺及多发淋巴结转移。详见表1。

表1 11例头颈部非横纹肌肉瘤软组织肉瘤患儿临床特征Tab.1 Clinical features of 11 children with head and neck non-rhabdomyosarcoma soft tissue sarcoma
2.2 治疗情况及近期疗效
5例患儿行手术治疗,6例患儿仅行活检。全部患儿均接受化疗。3例患儿行外放疗,4例患儿未放疗,4例患儿行125I粒子植入治疗。随诊时间10~78个月,中位随诊时间19个月。8例患儿达到完全缓解,1例患儿因疾病进展死亡,2例患儿带瘤生存,其中1例为部分缓 解,另1例发生肺转移正在治疗中,详见表2。
2.3 进展及复发情况分析
生存分析结果显示:本组患儿预计平均生存时间为(64.7±6.9)个月(95%CI:51.1~78.2)。1年总生存率及无事件生存率均为(88.9±10.5)%。治疗过程中进展2例,其中1例患儿初诊时活检病理提示为婴儿型纤维肉瘤,给予化疗3个疗程后肿瘤进展,行手术切除后,病理诊断为滑膜肉瘤,更改化疗方案为VDC/IE方案交替,目前随诊14个月,出现肺转移,已经行肺转移瘤灶切除术,仍在治疗中。另1例为颈部恶性横纹肌样瘤患儿,治疗中局部进展,予125I粒子植入治疗后达到局部控制,目前随诊72个月,为完全缓解状态。死亡患儿1例,为恶性横纹肌样瘤,初诊时即存在远处转移,予化疗6个月,肺部转移瘤灶进展死亡。详见表3。
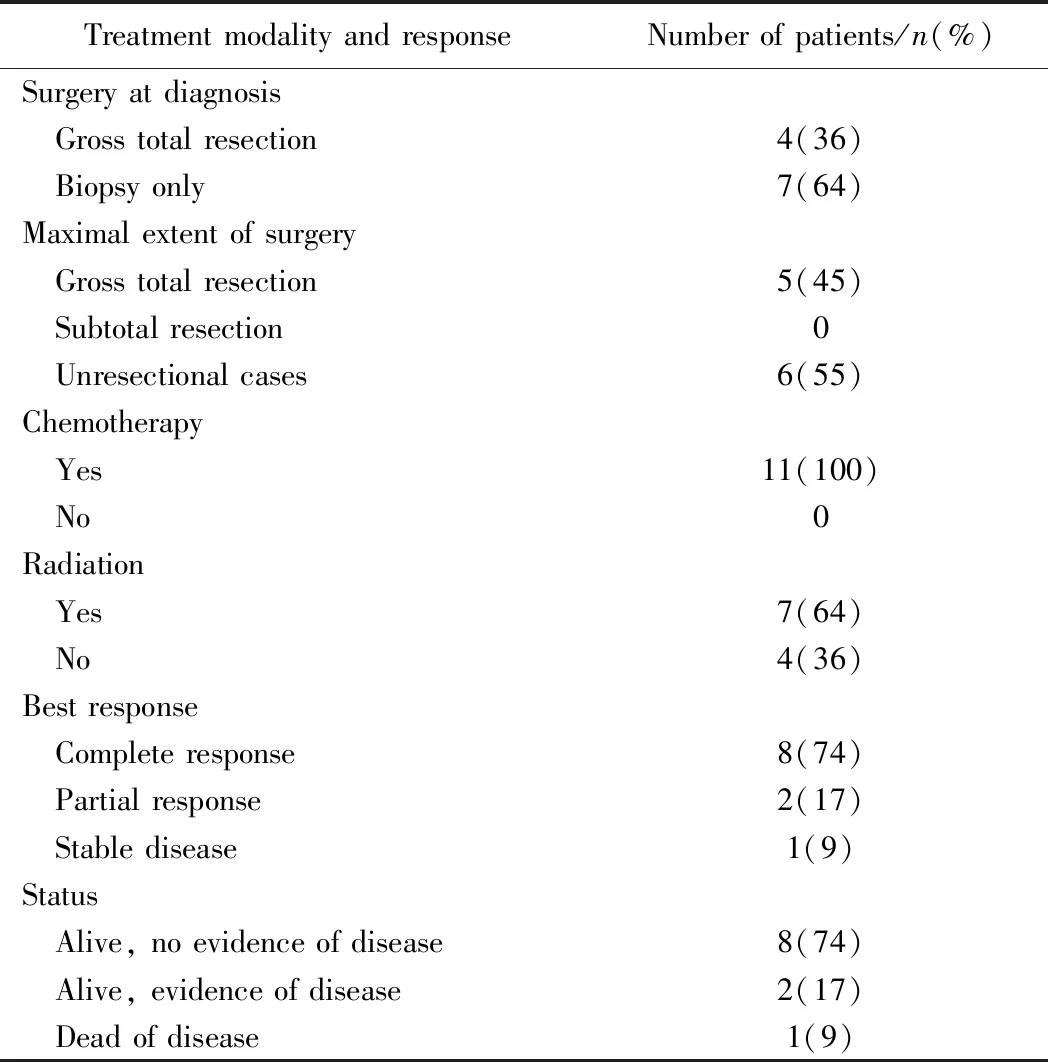
表2 11例头颈部非横纹肌肉瘤软组织肉瘤患儿治疗情况及预后Tab.2 Treatment and response of 11 children with head and neck non-rhabdomyosarcoma soft tissue sarcoma
2.4 治疗相关不良反应
本组患儿未见严重骨髓抑制伴致死性感染发生,未见Ⅲ~Ⅳ级脏器功能损害。放疗或125I粒子植入后未见严重皮肤黏膜毒性反应发生。
3 讨论
儿童及青少年头颈部非横纹肌肉瘤软组织肉瘤是一组罕见的肿瘤,国内尚无关于本病的大样本的回顾性研究。本研究是对本中心近5年来收治的头颈部NRSTS患儿的回顾性总结。本单中心研究显示,头颈部的软组织肉瘤,病理类型多样,包括滑膜肉瘤、恶性横纹肌样瘤、纤维肉瘤及罕见的CIC-DUX4易位小圆细胞肉瘤,部位分布于头面部软组织、口腔、脑膜旁区及颈部。肿瘤直径>5 cm的占55%,局部淋巴结的转移发生率为45%,远处转移的发生比例为36%。全身化疗联合手术及放疗是有效的治疗手段,通过多学科综合治疗,9例(91%)患儿均达到了完全缓解或部分缓解,近期治疗效果较理想。
非横纹肌肉瘤软组织肉瘤的组织亚型有50多种,异质性强,其临床表现及治疗效果差异较大。据报道[6-12]儿童NRSTS约占儿童软组织肉瘤的一半,病理类型主要为:滑膜肉瘤、恶性周围神经鞘瘤、恶性纤维组织细胞瘤和纤维肉瘤。NRSTS的危险度分组参照美国COG指南推荐标准,所有低级别肉瘤或直径小于等于5 cm的高级别肉瘤属于低危组;直径>5 cm的高级别肉瘤或有肉眼残留者为中危组;确诊时发生远处转移者为高危组[13-15]。肿瘤组织学等级较高、侵袭性强,肿瘤直径>5 cm,及手术不能完整切除是影响NRSTS预后的主要因素。
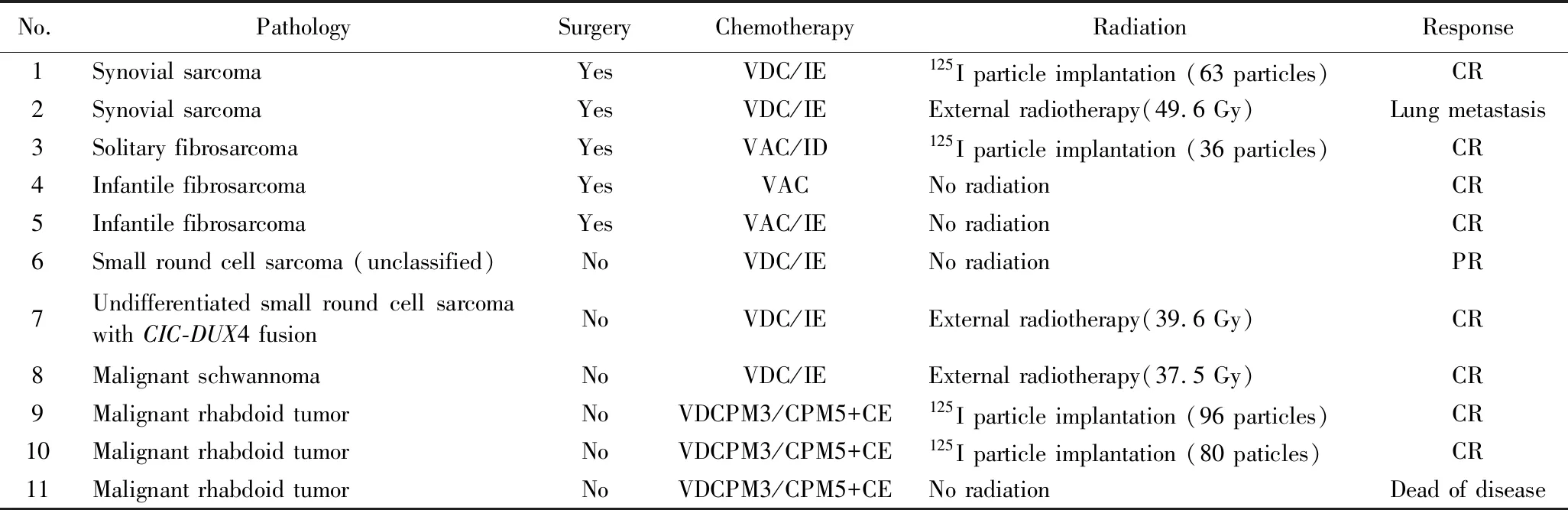
表3 11例头颈部非横纹肌肉瘤软组织肉瘤患儿治疗情况Tab.3 Treatment modality of 11 children with head and neck non-rhabdomyosarcoma soft tissue sarcoma
来自美国圣吉德儿童医院对于NRSTS 的回顾性研究[2]显示,头颈部NRSTS局部控制率及总体生存率较发生于除内脏部位的其他部位的NRSTS低。在成人患者,头颈部NRSTS约占软组织肿瘤的10%,治疗为包括手术、化疗及放疗在内的综合治疗。与儿童的研究结果相似之处为,疾病控制率在仅手术组,手术联合放疗组是一致的。此外,尽管多种化疗药物应用于成人包括头颈部在内的高危软组织肉瘤,化疗的效果有限,圣吉德儿童医院的研究[2]显示,辅助化疗对于生存率的影响是有限的。尽管如此,本研究结果显示,化疗仍是重要的全身治疗手段,本组患儿中4例高度恶性软组织肉瘤,包括2例恶性横纹肌样瘤,1例CIC-DUX4易位小圆细胞恶性肿瘤及1例恶性外周神经鞘瘤通过化疗联合外放疗或125I粒子植入治疗达缓解。
恶性横纹肌样瘤为高度恶性、进展迅速的肿瘤,多数患儿在诊断后12个月内发生死亡。美国肾母细胞瘤协作组统计的数据[16]显示,恶性横纹肌样瘤总的生存率仅为23.2%。颅外及肾外恶性横纹肌样瘤更为罕见。本组患儿中包括恶性横纹肌样瘤患儿3例,其中2例患儿经过化疗联合125I粒子植入治疗得到了长期缓解,1例患儿因早期存在远处转移,治疗效果不佳。
外科手术是重要的治疗手段,目前手术方式的改进已经在最大程度上体现了功能的保留。肺转移灶的手术切除目前也普遍应用到非横纹肌肉瘤软组织肉瘤的综合治疗中。有报道肺转移瘤经过手术切除,中位生存期可以增加2~125个月[17]。本研究中,1例滑膜肉瘤患儿,前期经过化疗、手术及局部放疗,治疗1年后出现肺部转移,已行肺转移瘤灶切除,目前仍在巩固治疗中。放疗是重要的局部控制手段,一项研究[18]显示,Ⅱ期患儿术后予以辅助放疗5年局部控制率82%,较未放疗组的43%有明显提高。
分子生物学技术已经广泛应用于软组织肉瘤辅助诊断及分型中[19]。国内外的数据[1,10]显示,儿童最常见的病理类型为滑膜肉瘤(synovial sarcoma, SS),约占到非横纹肌肉瘤软组织肉瘤的30%~35%。滑膜肉瘤存在t(x; 18)(p11,q11)基因的融合,导致SYT-SSX基因的融合,该融合基因的检测已经被认为是滑膜肉瘤诊断的金标准[20]。本研究中2例滑膜肉瘤患儿均存在SYT-SSX基因的融合。婴儿型纤维肉瘤(infantile fibrosarcoma, IF)是发生在5岁以内婴幼儿的纤维肉瘤,预后较好,研究[21]显示,婴儿型纤维肉瘤有染色体11、20、17和8的增加。具有诊断意义的染色体异位为t(12;15)(p13;q25),导致ETV6-NTRK3融合基因[21]。
目前仍有一些小圆细胞软组织肉瘤不能分类,t(4;19)(q35;q13.1)易位,导致染色体19q13.2上的CIC和染色体4q35.2上的DUX4融合。最新的研究[22]已经表明CIC-DUX4易位肿瘤是一种新的罕见的易位相关的软组织肉瘤,具有特异的组织病理学特征,该肿瘤为高度侵袭性,发展迅速,国外研究[22]报道最长生存时间不超过16.8个月。本研究中1例患儿经活检确诊为CIC-DUX4易位小圆细胞恶性肿瘤,该患儿对化疗十分敏感,经过化疗后肿瘤明显缩小,后予放疗局部控制,目前随诊19个月,为缓解状态。
总之,儿童头颈部非横纹肌肉瘤软组织肉瘤是一类罕见的、异质性较强的恶性肿瘤,需要包括手术、化疗及放疗在内的综合治疗;预后与病理类型、组织学分级、肿瘤大小、是否发生远处转移密切相关。由于头颈部特殊解剖结构的限制,部分患儿通过化疗联合外放疗或125I粒子植入治疗,可达到长期缓解。随着肿瘤免疫及分子靶向治疗研究的进展,近年来的临床试验,在传统化疗的基础上加入靶向药物,取得了一定的治疗效果。如西罗莫司和依维莫司对于血管周围上皮样细胞肿瘤和复发的淋巴管肌瘤或血管肌脂瘤的治疗获得令人满意的疗效。克唑替尼对于ALK基因异位的炎性成肌纤维细胞肿瘤有很好的疗效,贝伐单抗单药或与替莫唑胺联合方案在晚期或者复发转移的类上皮血管外皮细胞瘤和孤立纤维肿瘤耐受良好且有一定的疗效。伊马替尼已被批准治疗胃肠道间质瘤等。靶向药物的应用有望进一步提高儿童及青少年软组织肉瘤的治疗效果。
猜你喜欢
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
中国医学影像学杂志(2018年9期)2018-10-17 01:27:04
中国自行车(2018年8期)2018-09-26 06:53:34
中成药(2017年4期)2017-05-17 06:09:52
中华老年口腔医学杂志(2016年2期)2017-01-15 14:24:54
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:36
中国卫生标准管理(2015年14期)2015-01-27 02:24:27
西南军医(2014年4期)2014-03-03 16:50:35
