
乳腺癌中基于开放染色质的环状RNA互作网络构建及功能分析
2019-12-23 08:00张蕴显王雅梅
首都医科大学学报 2019年6期
张蕴显 王雅梅 周 萍*
(1.首都医科大学生物医学工程学院生物医学信息学系,北京 100069;2.首都医科大学基础医学院生物化学与分子生物学系,北京 100069)
女性乳腺由皮肤、纤维组织、乳腺腺体和脂肪组成。乳腺癌是一种发生在乳腺上皮组织中的恶性肿瘤。目前,乳腺癌已成为威胁女性身心健康的常见肿瘤。据估计,2017年美国有255 180例新发病例和41 070例乳腺癌死亡病例[1]。乳腺癌常见的3种亚型是雌激素受体阳性(estrogen receptor positive,ER+)、人类表皮生长因子受体2阳性(HER2+)和三阴型。相关研究[2-5]表明肥胖、雌激素和孕激素的使用、高龄初产、酗酒等因素能够增加乳腺癌的发生风险。除了以上几种因素外,基因突变、表观遗传机制在乳腺癌的发生、发展、转移中也起到重要作用。过去几年中,很多研究[6-8]表明非编码RNA(non-coding RNA,ncRNA)参与到致癌过程中,例如微小RNA(micro RNA,miRNA),长非编码RNA(long non-coding RNA,lncRNA)等[6],可以作为乳腺癌识别、预后的生物标志物[7]。作为新的内源性的ncRNA,环状RNA(circular RNA,circRNA)对肿瘤发生发展过程的调控作用已经成为研究热点之一[8]。
circRNA是一种新的内源性ncRNA,其长度为几百到几千个核苷酸[9]。与线性RNA转录产物不同的是,circRNA通过RNA的3′端和5′端的共价键形成,具有稳定的结构[10-11],但circRNA的生物发生过程尚不清楚。尽管如此,越来越多的研究[6]证明circRNA参与到了一系列的病理发生过程中。circRNA参与疾病发生过程的机制有以下几种途径:①circRNA作为竞争性内源RNA(competing endogenous RNAs,ceRNA)和“海绵吸附miRNA”(miRNA sponges)参与致癌的调节过程。miRNA通过与mRNA的3′端结合负向调节mRNA,有关研究[12]显示,其他具有miRNA靶点的RNA也同样可以与mRNA竞争miRNA。研究[13]显示含有miRNA的反应元件(miRNA reponse element,MER)的circRNA可以与miRNA相互作用并充当“海绵吸附miRNA”,以此削弱miRNA对靶基因的抑制作用。②circRNA可以特异性结合蛋白质,直接或通过RNA间接阻断蛋白质的功能[14-15]。③调节基因转录,虽然大部分circRNA作为ceRNA和“海绵吸附miRNA”调节miRNA,但相关研究[16]表明,一些circRNA可以顺式或反式的调节基因的转录。④普遍认为circRNA是一种特殊的内源性的ncRNA,不能直接翻译为蛋白质。但最近的研究[17-18]显示,一些动物体内circRNA(如小鼠、果蝇)可以直接翻译蛋白质并调控其生理过程。circRNA在乳腺癌患者体内存在差异表达,可能是乳腺癌细胞增生的生物标志物且与乳腺癌亚型相关[19-20]。
转座酶可接近性染色质测序技术(assay for transposase accessible chromatin with high-throughput sequencing, ATAC-seq),可以只利用少量细胞快速得到调控的多维信息。ATAC-seq可以同时获得“开放”染色质的位置、转录因子的结合位点、核小体的调控区域和染色质状态等信息,在表观遗传机制研究领域有广阔的应用前景[21-22]。脱氧核苷酸酶Ⅰ超敏感位点测序技术(DNase Ⅰ hypersensitive sites sequencing,DNase-seq)是利用脱氧核苷酸酶Ⅰ(DNase Ⅰ) 超敏感位点捕获技术,通过DNase Ⅰ对裸露在蛋白质包裹之外的DNA进行酶切,捕获基因活化区域DNA片段,然后进行高通量测序[23]。
本文利用ATAC-seq和DNase-seq数据,全基因范围分析乳腺癌MCF-7、T-47D和MDA-MB-231细胞系中circRNA的转录调控,进而构建了乳腺癌中活化circRNA基因的功能互作网络,从而挖掘出乳腺癌中的主要功能组circRNA和关键活化circRNA基因。
1 材料与方法
1.1 数据
本研究使用到乳腺癌的MCF-7、T-47D、MDA-MB-231细胞系的ATAC-seq和DNase-seq测序数据和人类circRNA数据库。①MCF-7、MDA-MB-231细胞系的ATAC-seq实验数据下载于基因表达数据库(Gene Expression Omnibus, GEO)(网址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97583)的GSE97583[24]实验数据。②MCF-7、T-47D细胞系的DNase-seq实验数据下载于UCSC基因浏览器中Table Browser的Duke DNase-seq HS实验数据(网址:https://genome.ucsc.edu/cgi-bin/hgTables),和GEO数据库的GSE108167(网址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108167)实验数据。③circRNA的高通测序数据下载于circBase中所有已发现的人类circRNA[25-27]实验数据(网址:http://www.circbase.org/)。
1.2 软件
本文所用软件有:Windows 10,Access 2010数据库,Cytoscape 3.6.1(网址:https://cytoscape.org/)版本的图形显示软件[28],Cytoscape3.6.1插件ClueGo V2.5.2[29],KEGG pathway数据库(网址:https://www.kegg.jp/kegg/mapper.html)。
1.3 方法
本文工作主要环节为特异circRNA筛选、网络构建和功能分析,方法流程图如图1所示。

图1 分析方法流程图Fig.1 The flow chart of analyzing method
1.3.1 于Access数据库的数据挖掘
首先利用Access 2010构建“乳腺癌开放染色质高通测序数据库”,该数据库基础表由6个高通测序样本、人类基因hg19(网址:http://genome.ucsc.edu/)和circRNA(网址:http://www.circbase.org/)构成,基因版本配准采用在线转换工具:http://genome.ucsc.edu/cgi-bin/hgLiftOver。然后利用SQL语句对数据进行筛选挖掘。数据筛选实例详见图2(T-47D的DNase-seq实验数据数据和circRNA数据为例)。

图2 数据筛选实例Fig.2 Examples of data mining
以此初步筛选出与乳腺癌相关的circRNA,以备进一步分析。
1.3.2 活化circRNA基因KEGG pathway富集筛选
将筛选出的活化circRNA对应的基因放入KEGG数据库中,利用KEGG Mapper对这些特异基因进行功能分析,并进一步筛选出与肿瘤(特别是与乳腺癌)相关通路的活化circRNA基因。在此结果中最终筛选出3个细胞系共有活化circRNA对应的基因,依据Gene Ontology(GO)的分子生物过程构建乳腺癌活化circRNA基因互作网络。
1.3.3 基于GO生物过程的乳腺癌活化circRNA基因互作网络构建
在Cytoscape 3.6.1平台中,使用ClueGo 2.5.2将经过数据挖掘筛选出的活化circRNA基因的集合和KEGG pathway富集筛选出的3个细胞系共有的活化circRNA基因的集合依据GO 生物过程构建乳腺癌活化circRNA基因互作网络,GO版本为Biological Process-EBI-UniProt-GOA(日期:2018-04-09),网络构建方法及参数选择:依据GO 术语进行分组,网络特异性为中等,P<0.05,为了细化模组功能,GO 术语层级设置为10~20级,模组最少基因数为3,占所在功能或通路基因的百分比为4%以上,网络连接度的Kappa 评分为0.5,富集分析为双尾超几何分布检验,P值校正方法为“Bonferroni step down”。
2 结果
2.1 MCF-7高敏感位点circRNA构成比
本文分析MCF-7细胞捕获高敏感位点circRNA为7 458个,含有utr区的基因是10 163个,捕获不含3utr和5utr的基因的circRNA是3 501个。由此可见在乳腺癌中circRNA的表达调控是非常活跃的。根据circRNA综合数据库CIRCpedia v2(网址:http://www.picb.ac.cn/rnomics/circpedia/)的研究,线性RNA中也包含一定比例的circRNA[30]。为此本研究以DNase-seq和ATAC-seq所有捕获的circRNA为研究对象,即不考虑该基因是否包含utr区。
2.2 3种乳腺癌亚型对比
T-47D捕获circRNA最高,其次是MCF-7,最少的是MDA-MB-231。就两种实验而言,DNase-seq捕获率远高于ATAC-seq。将3种细胞捕获的活化circRNA基因分别构建了互作网络,从结果看它们拓扑结构基本相似,只是每个功能模组基因构成比例略有差异。
综上所述,莱考夫非常强调概念隐喻在数学认知中的作用,认为概念隐喻是理解复杂数学思想的核心认知机制。概念隐喻是算术所需要的一种重要的认知能力。人们正是通过隐喻对数学中的概念进行认知和理解,概念隐喻使人们超越了极少的先天算术和简单的数数能力,延伸了人们的认知能力,获得了进一步的算术能力。物体集合隐喻、对象建构隐喻、量尺隐喻和沿路线运动隐喻是四种基本的基础隐喻,也是人们扩展算术的重要的隐喻能力。这四种基本的基础隐喻都是以隐喻描述了人们的日常经验与数字之间的映射关系。
本研究结果显示,3种细胞捕获的活化circRNA基因主要富集在细胞代谢的生物过程中,其中DNase-seq实验捕获的MCF-7和T-47D细胞的活化circRNA基因富集在有机氮化合物代谢过程(分别占11.58%、10.67%);ATAC-seq实验捕获的MDA-MB-231和MCF-7细胞的活化circRNA基因分别主要富集在细胞大分子代谢过程(15.43%)和细胞蛋白质代谢过程(18.35%)。
除了细胞代谢的生物过程,本研究捕获的活化circRNA基因也显著富集在其他与肿瘤相关的生物过程中,包括:DNase-seq实验捕获的MCF-7细胞的活化circRNA基因显著富集在细胞正调节(7.61%)、细胞内转导(3.65%)、细胞组分生物合成调控(3.57%)、生物过程负调控(3.01%)、细胞有丝分裂周期(2.3%);DNase-seq实验捕获的T-47D细胞的活化circRNA基因显著富集在细胞正调节(7.26%)、超分子纤维组织(3.48%)、细胞蛋白质定位(3.63%)、生物过程负调控(2.74%)、细胞有丝分裂周期(2.3%);ATAC-seq实验捕获的MDA-MB-231细胞的活化circRNA基因显著富集在细胞组分组织正调节(10.8%)、细胞分解代谢过程(8.49%)、细胞内转导(4.94%)、细胞骨架组织(4.01%)、蛋白质定位(3.24%)、细胞有丝分裂周期(3.09%);ATAC-seq实验捕获的MCF-7细胞活化circRNA基因显著富集在细胞过程正调节(9.8%)、细胞器蛋白质定位(9.24%)、细胞蛋白质定位(5.6%)、细胞组分装配(4.06%)、蛋白质定位(2.94%)、有丝分裂细胞周期过程(2.8%)。为此本研究用3种细胞系共有的circRNA进行进一步研究。
2.3 KEGG功能分析
对3种细胞捕获的活化circRNA基因进行KEGG功能分析,由结果可见比对到的关键信号通路皆与肿瘤有关。本研究中选取了对肿瘤起关键作用的18个信号通路(表1)。根据这18个信号通路最终筛选出了326个共有活化circRNA基因,并据此进行共有基因的表达调控互作网络构建及GO分子功能富集分析。
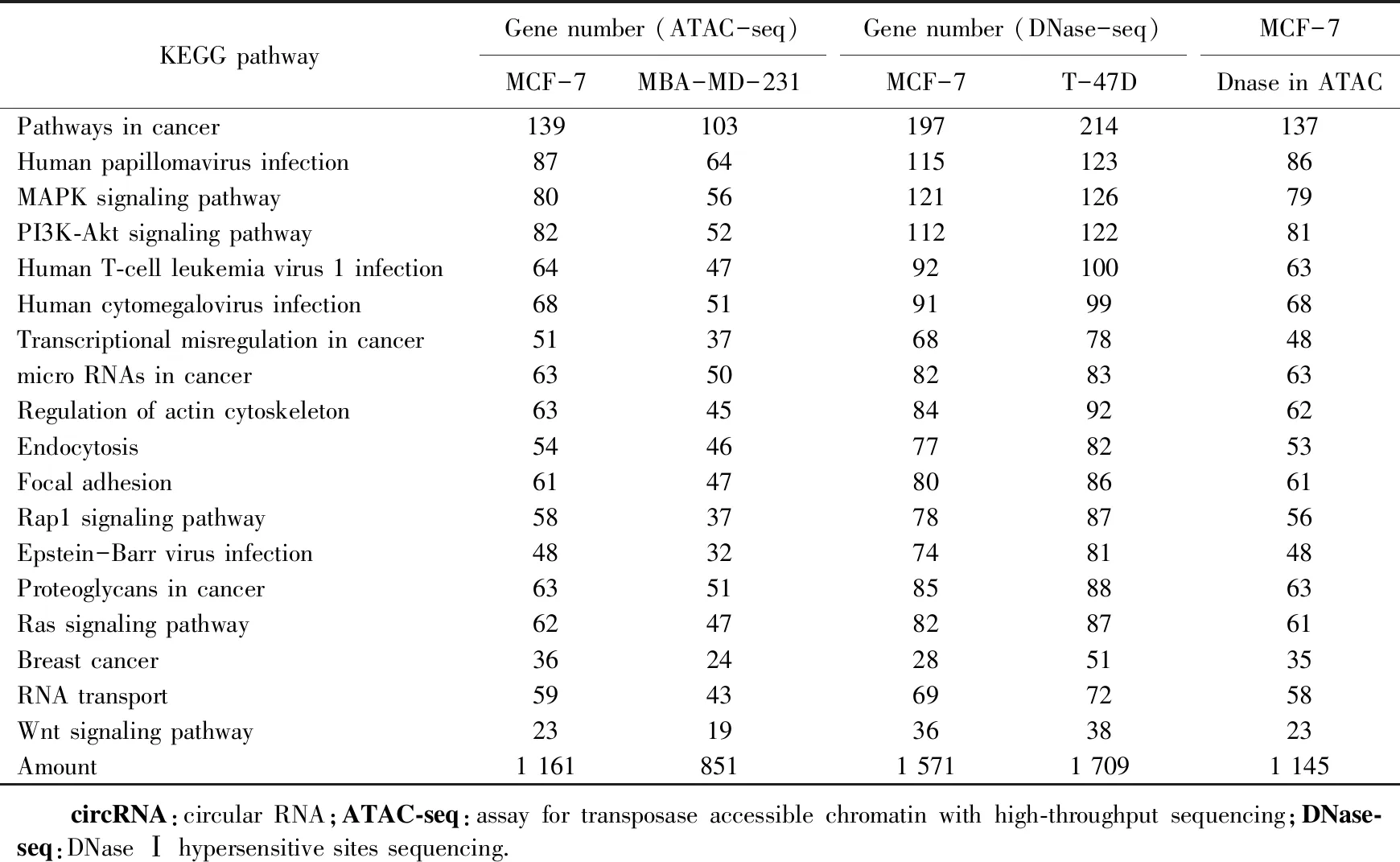
表1 活化circRNA基因所属关键信号通路Tab.1 Key signaling pathways for activated circRNA gene
2.4 互作网络构建及分子功能分析
为了全面探索3个细胞系共有活化circRNA基因的生物学功能,本研究进行分子功能富集分析及生物过程的表达调控互作网络构建。分子功能富集分析结果如图3所示,由结果可见,这些活化circRNA基因显著富集在结合蛋白质和刺激活化功能区。

图3 活化circRNA基因分子功能Fig.3 Molecular function of activated circRNA gene
利用Cytoscape 3.6.1的ClueGo插件构建的生物过程表达调控互作网络结果如图4所示。
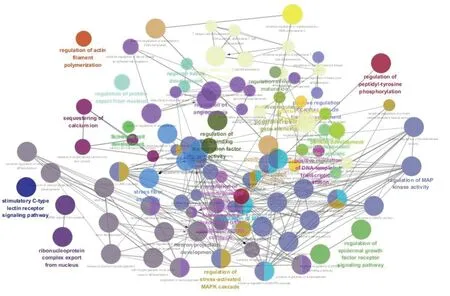
图4 活化circRNA基因功能互作网络Fig.4 Functional interaction network of activated circRNA genes
根据构建的生物过程表达调控互作网络,其功能模组主要分布于细胞代谢、重要信号通路、生物过程的调控、生物过程反应等生物过程中,结果如图5所示。根据主要的功能模组最终本研究识别出了PTK2B、PDCD6IP、ABL1、EGFR、RHOA、MTOR、NRP1、ATR、CTNNB1、ILK、NF1、PRKCA、HNRNPK、MEF2C、PMAIP1、LYN、NR4A3、SRF、AREG、PML、PTK2、ROCK2、TGFBR2、VEGFA、DLG1、HRAS、ITGA2、CREB1、STAT3、RUNX2、TIAM1等31个hub基因。这些基因所对应的捕获到的活化circRNA如图6所示。

图5 活化circRNA基因重要功能组Fig.5 Important functional groups of activated circRNA genescircRNA:circular RNA.
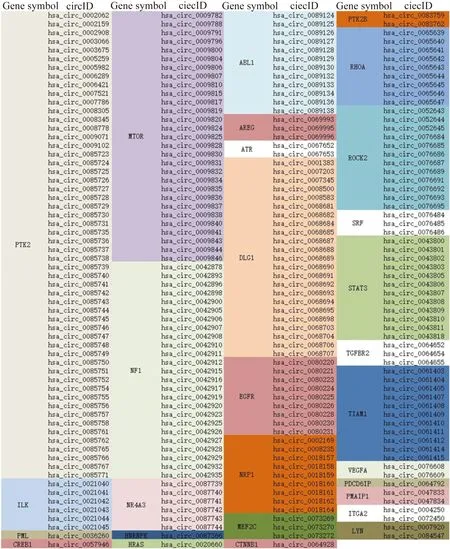
图6 hub基因的活化circRNA列表Fig.6 Activated circRNA list of hub genescircRNA:circular RNA.
从图6中看出,PTK2基因对应的活化circRNA最多,PTK2在ErbB信号通路中,对细胞粘连迁移起直接作用,由于PTK2活化circRNA多,因此circRNA吸附的miRNA的数量和种类也会随之增多,从而降低了miRNA对PTK2基因的mRNA调控。其次是MOTR基因对应的活化circRNA,MOTR基因在乳腺癌通路中直接对细胞的增生和存活起作用。由此可见上述circRNA在乳腺癌中起重要作用。
由于circRNA可以直接影响其对应基因的蛋白质表达水平, circRNA活化程度越高其对应基因的蛋白质表达水平也会增高,图5中显示了依据活化circRNA构建的基因互作网络功能组,更显示出了circRNA在乳腺癌中的作用。
3 讨论
随着circRNA领域的发展,已经建立了许多关于circRNA的数据库以促进circRNA的分析。circBase,CIRC pedia v2和Deepbase 2.0数据库包含许多关于不同物种的circRNA,并提供详细信息。CSCD和CircInteractome数据库可用作预测miRNA反应元件和RNA结合蛋白质的工具。CirclncRNAnet数据库提供了一种分析测序结果的简便方法。 ExoRBase数据库提供了人类血液外泌体中存在的58 330个circRNA。 circRNA Disease数据库记录了多种疾病中经过实验验证的circRNA[31]。Starbase v2.0数据库是一个提供miRNA-ncRNA互作网络和生物信息学应用的数据库,该数据库的开发者利用108个CLIP-seq(PAR-CLIP,HITS-CLIP,iCLIP,CLASH)数据集鉴定出了9 000多个miRNA-circRNA互作关系[32]。最近有研究人员[33]使用Starbase v2.0数据库鉴定CLIP-seq数据中miRNA-circRNA的互作关系,并开发了circSeeker,circAnno和clipSearch生物信息学分析软件用于注释和识别circRNA及其与miRNA的相互作用。目前对circRNA生物信息学分析大多基于表达谱数据筛选在肿瘤细胞内差异表达的circRNA,并利用上述公开数据库和软件分析预测circRNA-miRNA的互作网络,例如有研究人员[34]利用表达谱数据筛选出了肝癌细胞中差异表达的circRNA,并利用CSCD和CircInteractome数据库预测差异表达的circRNA的潜在靶miRNA,构建circRNA-miRNA-mRNA互作网络,揭示circRNA对肝癌的调控作用。本研究立足于circRNA的表达调控,借助Access数据库使用SQL语句对MCF-7、T-47D、MDA-MB-231细胞系的ATAC-seq和DNase-seq实验数据进行分析挖掘,鉴定出在乳腺癌细胞中可能出现circRNA的活化染色质位点,筛选出3种乳腺癌共有的活化circRNA基因,并从功能角度构建活化circRNA互作网络。由于目前对circRNA的注释信息尚不完善,而对基因的注释比较完整,因此笔者利用ClueGo插件对活化circRNA基因进行生物学分析,并构建活化circRNA基因的生物过程调控互作网络,以此预测筛选出的活化circRNA对乳腺癌的调控作用。
目前相关研究[35]表明,癌细胞的一个新兴特点是改变代谢。肿瘤的发生发展依赖细胞代谢重编程,细胞代谢重编程也是致癌性病变的直接或间接结果[36]。本研究的结果显示,乳腺癌细胞染色质中处于活化状态的circRNA对应的基因,其参与的生物过程主要是细胞代谢,正、负调控,细胞周期,细胞凋亡、增生、分化,重要信号通路,细胞反应等重要的细胞生物过程,因此表明circRNA在乳腺癌的发生发展过程中起到重要作用。尤其是发现的hub基因的circRNA起到至关重要的作用。
本研究发现尽管一个基因编码区可能包含多个circRNA,但并非捕获到基因编码区上全部circRNA。例如PTK2B基因在其编码区内有8个circRNA:hsa_circ_0083760、hsa_circ_0083759、hsa_circ_0083765、hsa_circ_0083764、hsa_circ_0083761、hsa_circ_0083762、hsa_circ_0083758和hsa_circ_0083763,但在本研究中仅捕获到hsa_circ_0083759和hsa_circ_0083762,根据miRTarBase(miRNA靶基因预测数据库)数据库hsa_circ_0083759和hsa_circ_0083762可能作为hsa-miR-23b-3p、hsa-miR-517c-3p和hsa-miR-517a-3p的竞争性内源RNA和“海绵吸附miRNA”,这说明在乳腺癌中circRNA的启动是有针对性的,很可能是针对某些特定的miRNA作为竞争性内源RNA和“海绵吸附miRNA”。
在本研究中连接度最高的circRNA对应的基因是PTK2B,该基因编码细胞质蛋白质酪氨酸激酶。最新研究[37]表明,PYK2B水平可以预测结肠腺癌手术切除后的预后。各种治疗模型中circRNA的表达谱尚未得到广泛研究,但已经报道了在放射抗性癌细胞中cirRNA的差异表达[38]。虽然目前乳腺癌还没有广泛开展PYK2B基因的circRNA研究,但是本文研究表明,PYK2B基因的circRNA很可能对乳腺癌的诊断治疗起重要作用。最近的研究[39]已经证明外泌体介导肿瘤微环境中不同类型细胞之间的相互作用,它们调节肿瘤生长、转移、耐药性(通过转运肿瘤相关mRNA,miRNA和蛋白质)、血管生成、免疫逃逸和其他过程。PDCD6IP基因对外泌体的发生有着重要的调控作用,能直接介导肿瘤微环境中对肿瘤的重要调控作用。ABL1基因是一种蛋白质酪氨酸激酶和有效的致癌基因,相关研究[40]证实,它是miR-203(一种肿瘤抑制性miRNA)的真正靶标。ABL1基因通过与miR-203结合导致miR-203肿瘤抑制能力减弱或者丧失,使正常抑制的致癌基因不受表达,从而达到调控肿瘤发生发展和转移。虽然ABL1基因的circRNA对乳腺癌的作用目前尚未有相关研究,但ABL1基因的circRNA极有可能作为ceRNA和“海绵吸附miRNA”参与到乳腺癌致癌的调节过程。最近的相关研究[40]证明circHIPK3可以使内源性miR-7结合以隔离和抑制miR-7活性,从而导致表皮生长因子受体(epidermal growth factor receptor,EGFR)表达增加达到调控直肠癌生长和转移的作用[41]。虽然尚未有EGFR基因的circRNA对乳腺癌的作用的相关研究,但本研究证明EGFR基因的circRNA很可能对乳腺癌的诊断及预后有重要作用。RhoA表达水平在多种肿瘤组织中增加并且与肿瘤恶性相关,表明RhoA在肿瘤发生和转移中的重要作用。相关研究[42]证实miR-200b可介导RhoA基因以形成内源竞争并在包括肝细胞癌在内的多种肿瘤的发生中具有重要的调节作用。研究[42]显示,通过多种信号转导途径,RhoA参与肿瘤的恶性转化、侵袭、转移、血管生成等。RhoA基因的circRNA极有可能作为ceRNA和“海绵吸附miRNA”调控RhoA基因的表达并参与到乳腺癌致癌的调节过程中,并作为用于治疗癌症转移的潜在的治疗标靶。
本文从生物信息学角度对高通测序数据进行深度挖掘,发现的hubcircRNA基因及其对应的circRNA对乳腺癌基础临床研究具有一定的参考价值。
本研究通过3种乳腺癌细胞的DNase-seq数据和ATAC-seq数据对3种乳腺癌细胞内circRNA捕获率的分析,发现DNase-seq实验的捕获率及精度要高于ATAC-seq实验。通过对捕获的活化circRNA基因的生物信息学分析发现,乳腺癌细胞内的活化circRNA基因大多参与到细胞代谢,正、负调控,细胞的凋亡、增生、分化,以及目前已证实的跟肿瘤相关的一些重要信号通路中,因此表明活化circRNA在乳腺癌的发生发展过程中起到重要作用。本研究显示基因编码区包含多个circRNA,但在本研究中仅捕获基因编码区中对应的部分circRNA,表明这些基因对应的circRNA可能针对某些特定的miRNA作为ceRNA和“海绵吸附miRNA”参与到乳腺癌致癌的调节过程。通过对本研究识别出的hubcircRNA基因进行生物信息学分析,发现该基因参与到了肿瘤的恶性转化、侵袭、转移等过程中,这表明hubcircRNA在此过程中起重要作用,虽然目前尚未广泛研究这些hubcircRNA对乳腺癌的作用,但本研究的结果表明这些hubcircRNA可能作为用于治疗癌症转移的潜在靶点。
猜你喜欢
中老年保健(2022年6期)2022-08-19
材料与冶金学报(2022年2期)2022-08-10
昆明医科大学学报(2022年1期)2022-02-28
科学与财富(2021年33期)2021-05-10
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
中国生殖健康(2019年2期)2019-08-23
中国生殖健康(2019年6期)2019-01-06
