
非小细胞肺癌的新曙光:抗血管生成治疗联合免疫治疗
2019-12-20 01:26周建娅阮柯欣
浙江医学 2019年23期
周建娅 阮柯欣
近年来,肿瘤发病率和死亡率在全球范围内迅速增长,根据GLOBOCAN2018癌症报告,2018年全球癌症新发病例约为1 800万例,死亡病例约960万例[1]。肺癌年发病例约为210万例,年死亡病例约180万例,被称为肿瘤“第一杀手”,其中男性是肺癌发病率较高人群[1]。肺癌包括小细胞肺癌(small cell lung cancer,SCLC)和非小细胞肺癌(non-small cell lung cancer,NSCLC),其中SCLC约占15%,NSCLC约占85%。多数患者初次确诊时已属晚期,常因肿瘤转移而失去手术机会,而晚期肺癌患者5年生存率仅为4%[2]。近年来肺癌精准治疗发展迅速,使得表皮生长因子受体(EGFR)、间变型淋巴瘤激酶(ALK)和C-ROS原癌基因1酪氨酸激酶(ROS1)等驱动基因异常的肺癌患者有了相应的酪氨酸激酶抑制剂(TKI)可供治疗选择,但这些药物仅能使部分人群获益[3]。如EGFR基因突变在亚裔不吸烟女性患者中的比例约为50%,而在高加索人群中仅10%~20%的肺腺癌患者发生EGFR基因突变。免疫检查点抑制剂(以下简称免疫治疗)可通过选择性抑制程序性死亡受体1(PD-1)/程序性死亡配体 1(PD-L1)通路,激活杀伤 T淋巴细胞产生抗肿瘤活性。因其具有普适性及有效性,在肿瘤治疗领域迅速占据了重要地位,改变了多种恶性肿瘤的治疗模式,使部分晚期肿瘤患者获得了长期疗效。在KEYNOTE024临床研究中,采用帕博利珠单抗治疗晚期肺癌患者,其无进展生存期(PFS)为10.3个月,较传统化疗(6.0个月)明显延长,死亡风险也降低了40%,因此被美国食品药品监督管理局批准用于PD-L1高表达晚期NSCLC的一线治疗[4]。但仍有半数以上的患者难以从免疫治疗中获益,尤其是PD-L1低表达的人群。
通过出芽方式形成新生血管是肿瘤的特征之一,抗血管生成药物可以通过阻断新生血管的形成起到抗肿瘤治疗的作用,也可以逆转肿瘤微环境的免疫抑制状态,为免疫治疗打好基础。使用抗血管生成药物可以使肿瘤内血管存在一段“正常化”的时间窗,期间可加强免疫治疗药物及免疫细胞的运输,提高免疫治疗的疗效,从而增强抗肿瘤效果。因此,抗血管生成治疗联合免疫治疗有望成为一种新的治疗选择。
1 血管生成
血管生成是一个复杂的过程,受许多信号通路的调节,在胚胎的发育、成人的月经周期和病理性损伤后修复等活动中起着重要作用。虽然在生理过程中也存在血管生成,但是肿瘤的新生血管在功能和结构上与正常血管不同。
Folkman提出的出芽式血管生成模型被认为是肿瘤血管生成的主要形式。在出芽式血管生成模型中,内皮细胞通过改变表型转化为具有不同结构和功能的尖端细胞和柄细胞。它们的增殖、迁移和融合形成了新生血管腔。血管周细胞紧贴于内皮细胞之外,起着保持微血管稳定、调节内皮细胞功能等作用。由于血管生成因子和抗血管生成因子的失衡,导致了内皮细胞的大量增殖和迁移,异常增多的内皮细胞和血管周细胞形成了高通透性的管状和囊状血管。在肿瘤血管中,血管周细胞的低覆盖率导致了血管的高通透性,使得肿瘤血管内维持渗透压的小分子持续流失,这种压力梯度的减低最终影响了血流和大分子物质的运输,造成肿瘤组织的缺氧,也影响了抗肿瘤药物的运输及其作用效果[5]。因此,许多肿瘤通过影响血管生成机制来刺激肿瘤的生长,从而导致疾病进展。
血管内皮生长因子(VEGF)家族是调控血管生长的重要因子。在哺乳动物中,VEGF家族中有5种异构体,分别为 VEGF-A、VEGF-B、VEGF-C、VEGF-D 及胎盘生长因子(PLGF)。这些蛋白对应3种酪氨酸激酶受体,即VEGFR1、VEGFR2和VEGFR3。在血管生成的前端是VEGF浓度最高的地方,VEGF结合内皮细胞上的VEGFR2,促进尖端细胞和柄细胞表型转变,诱导尖端细胞迁移。VEGFR2激活下游ERK1/2信号通路后,受体将迅速清除配体并重新接受信号刺激。受体的这种迅速更新,有利于感受高浓度的VEGF并决定新生血管的走向[6]。
内皮细胞TEK酪氨酸激酶(Tie2)是一种在内皮细胞上高度表达的跨膜酪氨酸激酶受体,配体为血管生成素(Ang)1和Ang2。在内皮细胞中,Ang2与Tie2特异性结合后能抑制下游的信号通路,从而引起血管结构不稳定和新生血管对血管生成信号敏感性增强。在缺乏Ang1的未成熟肿瘤血管中,Ang2仍可通过激活Tie2上的整合素和下游信号通路促使尖端细胞的迁移[7]。
肝细胞生长因子(HGF)通过激活内皮细胞表面的间充质-上皮转化因子(c-MET)信号通路促进细胞的存活、扩增和上皮间质转化,加强细胞的运动和侵袭,促进血管生成,抑制内皮细胞凋亡[8]。目前已在多种肿瘤中发现,由于基因的异常扩增,转录的异常激活或缺氧等原因引起的c-MET及HGF过表达,且与不良预后相关[9]。
血小板衍生生长因子(PDGF)是由4种单链(PDGFA、PDGF-B、PDGF-C、PDGF-D)组成的二聚体,分别为PDGF-AA、PDGF-BB、PDGF-AB、PDGF-CC、PDGFDD。PDGF可诱导血管平滑肌细胞、结缔组织成纤维细胞、血管周细胞等间充质细胞的有丝分裂和趋化。激活PDGF/PDGFR通路可直接影响肿瘤的增殖,血管内皮细胞的重构、活化和血管周细胞的募集等[10]。
2 抗血管生成治疗
目前我国批准用于治疗晚期NSCLC的抗血管生成药物有贝伐珠单抗、重组人血管内皮抑制素和安罗替尼。
贝伐珠单抗是一种来自小鼠的人源化VEGF单克隆抗体,可特异性结合VEGF-A以阻断其与VEGFR1和VEGFR2间的相互作用。Reck等[11]一项关于贝伐珠单抗的Ⅲ期临床研究结果发现,虽然两组患者总生存期(OS)无明显差异,贝伐珠单抗剂量对疗效的影响也较小,但15mg/kg贝伐珠单抗联合吉西他滨/卡铂的高剂量组(HR=0.82,95%CI:0.68~0.98,P<0.05)、7.5mg/kg贝伐珠单抗联合吉西他滨/卡铂的低剂量组(HR=0.75,95%CI:0.62~0.91,P<0.01)的中位 PFS 均优于吉西他滨/卡铂对照组;药物有效率也明显提高(30.4%、34.1%比20.1%)。基于此研究,贝伐珠单抗被批准列入晚期NSCLC一线治疗。
重组人血管内皮抑制素是在人内皮抑素的N端加上9个氨基酸的衍生物,具有内皮抑素结合VEGF和FGF2阻断血管生成的作用,也能提高内皮抑素的稳定性,延长药物半衰期[12-13]。在一项Ⅲ期临床试验中,使用重组人血管内皮抑制素联合卡铂/长春瑞滨治疗晚期NSCLC患者,无论是在缓解率、临床获益率还是中位PFS上均优于安慰剂+卡铂/长春瑞滨的患者[14]。
安罗替尼是针对 VEGFR1-3、FGFR1-4、PDGFR、RET和c-Kit等多个靶点的TKI,具有显著的抗肿瘤效果[15]。ALTER0303是一项随机对照、双盲的Ⅲ期临床试验,结果显示接受安罗替尼为三线药物的晚期NSCLC患者可获得更长的中位PFS和OS(均P<0.01)[16]。
目前仍有众多针对NSCLC的抗血管生成药物处于早期临床试验阶段,如阻断HGF/c-MET通路的奥那妥珠单抗[17]、影响ANG/TIE2通路的Trebananib及多靶点的TKI[18]。但上述抗血管生成药物在短期有效后都会不可避免出现耐药,患者难以长期获益。因此,需要不断探索新的联合治疗策略,使患者获得长期受益。
3 免疫治疗当机体识别外来抗原时,
免疫系统会激活免疫反应对其进行清除。肿瘤细胞通过逃避抗原呈递细胞和常规T细胞对于肿瘤抗原的识别,从而抑制了毒性T细胞的激活。肿瘤通过募集调节T细胞等免疫检查点机制来逃避免疫系统对其的清除,从而在体内增殖扩散。许多研究证明,免疫检查点分子高表达的肿瘤患者往往预后不良。
PD-1是表达在NK细胞、B细胞、树突状细胞、单核巨噬细胞以及CD4+、CD8+T细胞表面的免疫检查点分子[19]。PD-1与表达在某些肿瘤细胞和免疫细胞上的配体PD-L1结合,可以抑制T细胞的激活、增殖,促进T细胞的凋亡[19-20]。激活PD-1/PD-L1通路可以诱导CD4+T细胞向调节性T细胞分化,调节性T细胞通过表达Foxp3转录因子等机制引起免疫抑制。PD-1/PDL1通路激活后,还可以诱导肿瘤细胞对T细胞介导的细胞毒性及其他抗肿瘤作用形成抵抗[21],从而产生耐药。抑制PD-1/PD-L1通路,可提高免疫细胞对肿瘤抗原的识别,激活CD8+T细胞对肿瘤细胞进行杀伤[22]。目前,阻断PD-1/PD-L1通路的抗PD-1抗体(如帕博利珠单抗、纳武利尤单抗)和抗PD-L1抗体(如阿特珠单抗、德瓦鲁单抗)已作为免疫检查点抑制剂,成为临床治疗NSCLC的药物[23]。
另一类免疫检查点分子表达在调节T淋巴细胞表面,以毒性T淋巴细胞相关蛋白4(CTLA-4)为代表。CTLA-4通过与抗原呈递细胞表面的CD80/CD86受体竞争性结合,阻断T细胞表达的CD28对抗原呈递细胞的激活,从而抑制免疫反应。调节T细胞还可以通过分泌IL-10等趋化因子来诱导效应T细胞的凋亡[24],从而形成免疫抑制。
免疫治疗的疗效在不同患者中的差异很大。目前研究发现肿瘤浸润淋巴细胞(TIL)可能与之相关。TIL是免疫治疗发挥抗肿瘤效应过程中不可或缺的参与者。肿瘤微环境的炎症状态大致可分为3类:大量的活性淋巴细胞浸润(热肿瘤)、淋巴细胞围绕在肿瘤边缘无法浸润到中心、没有淋巴细胞浸润(冷肿瘤)[25]。在有炎症反应存在的微环境中,免疫治疗对肿瘤的杀伤性是最大的。因此,如何联合其他抗肿瘤治疗改变患者免疫表型,使其获益于免疫治疗是进一步探索的方向。
4 联合治疗的基础
抗血管生成治疗和免疫治疗的抗肿瘤效果与肿瘤所处的微环境息息相关[26]。在如何提高免疫治疗效果的探索上,许多联合治疗策略着重于提高淋巴细胞对肿瘤的浸润,进而改变肿瘤微环境的免疫抑制状态,而抗血管生成治疗就是其中之一[27]。
4.1 VEGF的免疫抑制 VEGF是血管生成过程中的关键因子,同时具有免疫抑制作用,故使用VEGF相关抑制剂可以在抗血管生成层面上进行抗肿瘤治疗,也可以通过以下几个方面逆转肿瘤微环境的免疫抑制状态,为免疫治疗打好基础。
VEGF作用于树突状细胞表面的VEGFR2,通过抑制NF-κB通路阻止树突状细胞的成熟,进而阻止肿瘤的抗原呈递,导致肿瘤潜在的免疫逃逸。在许多恶性肿瘤患者中,高浓度VEGF与树突状细胞的缺乏或成熟障碍有关。通过对比接受贝伐珠单抗治疗患者治疗前后的血液样本,发现治疗后外周血中树突状细胞数量明显增多[28]。
VEGF可以通过抑制胸腺中的造血干细胞向CD4+、CD8+T细胞分化而直接影响T细胞的功能。VEGF结合于T细胞表面的VEGFR2,不仅可以直接抑制T细胞的增殖和细胞毒性作用,还可以通过上调PD-1、CTLA-4、TIM3等免疫检查点分子来抑制T细胞活性[29]。VEGF与外周血中的调节T细胞相关,转移性肾透明细胞癌患者在使用舒尼替尼后,外周血中的调节T细胞数量较前明显减少,且获得了更长的OS[30]。VEGF可诱导肿瘤血管表达FAS抗原配体(FASL),引起在肿瘤中浸润的CD8+T细胞凋亡,从而为肿瘤的免疫逃逸提供条件[31]。
血管内皮高表达的ICAM-1、VCAM-1和选择素E可以协助免疫细胞在血管内皮上的黏附,并从血管移出进入微环境发挥杀伤肿瘤的作用。VEGF可通过下调内皮细胞的ICAM-1和VCAM-1或阻止它们在内皮细胞上聚集,从而抑制淋巴细胞在肿瘤中浸润[32]。
4.2 Ang2的免疫抑制 Ang2募集表达Tie2的单核细胞并通过与Tie2结合,刺激单核细胞分泌IL-10。IL-10可诱导调节T细胞在肿瘤中的浸润,抑制T细胞活性。在胶质母细胞瘤的小鼠模型中,使用Ang2和VEGF的阻断剂可增加肿瘤组织中T细胞的浸润且减少肿瘤相关巨噬细胞的数量,逆转免疫抑制状态[33]。Ang2还可以诱导M2型巨噬细胞表达PD-L1,从而抑制CD8+T细胞的杀伤作用[34]。
Ang2能介导趋化因子CCL2的分泌,诱导巨噬细胞在肿瘤中的浸润,促进肿瘤的转移[35]。在使用Ang2阻断剂后发现内皮细胞上VCAM-1表达增加,间接证明了Ang2诱导VCAM-1下调,以此来阻止免疫细胞的浸润[36]。
4.3 HGF的免疫抑制 HGF作用于树突状细胞表面的c-MET,可以抑制树突状细胞的抗原呈递功能,还可以诱导T细胞分化为调节T细胞,进而分泌IL-10抑制肿瘤的免疫反应。HGF虽然能促进树突状细胞向淋巴结的迁移,但诱导了T细胞上CTLA-4的表达,从而抑制CD8+T细胞的激活[37]。并且通过抑制CD8+T细胞分泌IFNγ、穿孔素1和颗粒酶B等,进一步影响CD8+T细胞的杀伤作用[38]。
HGF可上调血管内皮细胞上稳定素1的表达,优先介导调节T细胞的迁移。在使用封闭稳定素1的单克隆抗体后的黑色素瘤小鼠模型中观察到,肿瘤组织中的调节T细胞、M2型肿瘤相关性巨噬细胞等免疫抑制细胞明显减少,并伴随着肿瘤生长受限和转移能力减弱[39]。由此可推测HGF通路可能与免疫抑制细胞的选择和肿瘤转移有关。
4.4 PDGF的免疫抑制 已有研究表明PDGFAB同样可抑制树突状细胞的成熟。PDGFAB还可以上调未成熟树突状细胞表面表达的CLEC2受体,该受体与T细胞表面配体结合后可诱导调节T细胞的生成[40]。PDGFBB被证实可促进肿瘤组织间充质干细胞的增殖[41],而间充质干细胞具有免疫抑制作用,进一步促进肿瘤的生长和免疫逃逸。
4.5 免疫反应对血管生成的影响 肿瘤的血管生成可引起微环境的免疫抑制,相应的肿瘤免疫对于肿瘤血管生成也有一定影响。在抗VEGFR2治疗耐药的胰腺癌和乳腺癌小鼠模型中,发现肿瘤组织表达的PD-L1水平异常增高[42]。进一步研究发现内皮细胞的PD-L1对于血管生成有一定的调节作用。在体外实验中,VEGFR2表达的增加与PD-L1的减少有关。这表明血管生成可能受到血管生成因子以外的细胞因子调节,同时这些细胞因子还可能与抗血管生成药物的耐药有关。
活化的CD8+T细胞分泌IFNγ作用于肿瘤基质中的受体1(IFNγR1)所引起的抗血管生成作用,可能是CD8+T细胞介导肿瘤清除的机制之一。IFNγ可以直接作用于内皮细胞,也可引起肿瘤相关成纤维细胞分泌的VEGF减少,从而导致血管的正常化或退化[43]。
5 联合治疗的临床应用
免疫细胞分泌的IFNγ促进血管正常化,正常化的血管又可在肿瘤中运输更多的免疫细胞和抗肿瘤药物,由此可推测,免疫治疗联合抗血管生成治疗在抗肿瘤治疗中具有增效作用。
关于抗血管生成治疗联合免疫治疗的临床应用探索越来越多。Reck等[44]一项随机、开放的Ⅲ期临床试验将1 202例转移性NSCLC患者按照1∶1∶1的比例分成3组,分别为在卡铂/紫杉醇化疗的基础上联合阿特珠单抗(ACP组,402例)、贝伐珠单抗(BCP组,400例)以及阿特珠单抗、贝伐珠单抗(ABCP组,400例),比较3种治疗方案给患者带来的临床收益。结果显示在无EGFR突变的野生型患者中,无论PD-L1是否高度表达,ABCP组在中位PFS(8.4个月比6.8个月;HR=0.59,95%CI:0.50~0.69)和中位 OS(19.8 个月比 14.9 个月;HR=0.76,95%CI:0.63~0.93)上均优于 BCP 组,而药物不良反应发生率比较差异无统计学意义;在EGFR基因突变及肝转移的亚组中,ABCP组中位生存时间仍明显优于BCP组。这项试验直接证明了免疫检查点抑制剂联合抗血管生成药物在转移性NSCLC中具有增效作用。
Rizvi等[45]选取了接受一线铂类化疗满4周期后42d内肿瘤无进展的NSCLC患者,进一步研究纳武利尤单抗联合贝伐珠单抗、单用纳武利尤单抗作为后续治疗的临床效果。患者被随机分入接受纳武利尤单抗(5mg/kg,3周/次)+贝伐珠单抗(15mg/kg,3周/次)治疗组和单用纳武利尤单抗(3mg/kg,2周/次)治疗组,直至疾病进展或患者发生不可耐受的不良反应。结果显示联合治疗组中位PFS为37.1周,明显优于单药治疗组(鳞癌患者16周,非鳞癌患者21.4周)。虽然联合用药的药物不良反应发生率(92%)较单药治疗(62%)有所增高,但均未发生4级及以上的严重不良反应。
目前,抗血管生成药物联合免疫检查点抑制剂在NSCLC中的临床试验正在开展。以下为登记在Clinical Trail网站上的、近期针对该类药物所开展的部分临床试验,见表1。
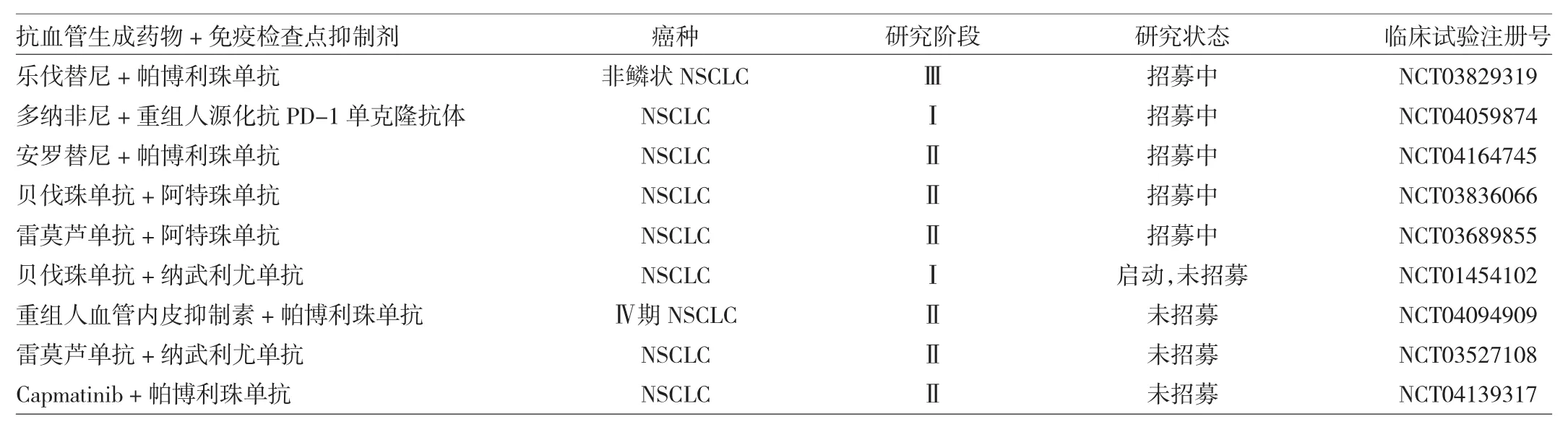
表1 近期针对抗血管生成药物联合免疫检查点抑制剂开展的部分临床试验
6 展望
越来越多研究表明抗血管生成联合免疫治疗可以使NSCLC患者获得更好的临床受益,但目前对其相关机制的认识仍是冰山一角。如何选择最佳的联合治疗药物方案,如何选择联合治疗的时间和最佳药物剂量,如何筛选可能获得最大受益的患者群体,如何选择对预后有影响的评价指标和生物标志物等等,都需要更多研究加以探讨。
猜你喜欢
中国循证儿科杂志(2022年3期)2022-12-15
中国肿瘤临床(2022年4期)2022-12-12
中国肿瘤临床(2022年21期)2022-11-27
中国现代医生(2022年19期)2022-11-04
广东海洋大学学报(2022年2期)2022-03-31
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
中华养生保健(2020年10期)2021-01-18
癌症进展(2020年23期)2020-12-22
临床肝胆病杂志(2020年2期)2020-12-14
中国生殖健康(2020年7期)2020-12-10
