
剑麻内参基因筛选与稳定表达分析
2019-12-19 02:13张燕梅王瑞芳杨子平鹿志伟李俊峰赵艳龙陆军迎周文钊
热带作物学报 2019年11期
张燕梅 王瑞芳 杨子平 鹿志伟 李俊峰 赵艳龙 陆军迎 周文钊

摘 要 為了筛选剑麻中稳定表达的内参基因,保证基因表达结果的可靠性,本研究以热麻1号为材料,采用实时荧光定量RT-PCR方法对来自剑麻转录组数据的8个内参基因(EF1α、MADH、ACT2、GAPDH、TUB、ACT54、CYP和EF1β)在烟草疫霉侵染、水杨酸(SA)、茉莉酸甲酯(MeJA)和脱落酸(ABA)处理过程中的表达水平进行检测,并结合GeNorm、BestKeeper和NormFinder软件综合评价8个内参基因的稳定性。结果表明:8个内参基因在不同处理下的表达稳定性存在显著差异,其中在烟草疫霉侵染和茉莉酸甲酯处理过程中,EF1α表达最稳定,EF1β表达最不稳定。在水杨酸处理过程中,EF1α表达最稳定,ACT2表达最不稳定。在脱落酸处理过程中,GAPDH表达最稳定,ACT54表达最不稳定。该结果为剑麻基因的表达分析提供了可供选择的内参基因。
关键词 剑麻;定量RT-PCR;内参基因
中图分类号 S563.8 文献标识码 A
Abstract In this study, eight reference genes (EF1α, MADH, ACT2, GAPDH, TUB, ACT54, CYP and EF1β) from the transcriptome database, were selected and analyzed in Rema No.1 including Phytophthora nicotianae Breda, MeJA, SA and ABA treatments. Statistical tools, including GeNorm, NormFinder and BestKeeper, were utilized to assess the suitability of reference genes based on the stability rankings for different species. The stability of the eight reference genes was significantly different under different treatments in Rema No.1. For Phytophthora nicotianae Breda and MeJA stresses, EF1α was identified as the most stable gene and EF1β was identified as the least stable gene. For SA stress, EF1α was identified as the most stable gene, and ACT2 was identified as the least stable gene. For ABA stress, GAPDH was identified as the most stable gene, and ACT54 was identified as the least stable gene. The results would provide reliable and optional available reference genes in gene expression analysis of sisal.
Keywords sisal; qRT-PCR; reference gene
DOI 10.3969/j.issn.1000-2561.2019.11.010
剑麻是我国热区特有的经济作物之一,剑麻不仅是主要的热带纤维原料[1],同时剑麻茎心是酿造龙舌兰酒的主要原料[2],剑麻麻渣含有较高皂素,可用来制药[3-4],剑麻也是一种重要的生物质能源[5],近年来,剑麻作为景天酸代谢的模式植物[6],有非常重要的理论和经济价值。
斑马纹病是剑麻的主要病害之一,主要由烟草疫霉引起。前期利用转录组测序从热麻1号中筛选了6849个差异表达基因[7],下一步将对部分候选基因进行表达研究,以期了解基因的功能和可能参与的信号途径。基因表达研究方法很多,其中qRT-PCR技术以其简单、快速、费用低且灵敏度高等优点成为目前研究的主要方法之一[8],由于不同样本RNA质量,反转录效率以及基因拷贝数等存在较大差异,在采用该方法时,需要一个表达相对稳定的内参基因来进行校正和标准化[9]。目前,龙舌兰属中常采用的内参基因有18SrRNA[7, 10]、ACT2[11]、Actin[12]、PU和UBQ11[13],由于基因表达受品种、不同器官以及环境条件影响较大[14-16],为了保证实验结果真实可靠,根据具体的实验条件,筛选适宜的内参基因十分必要。本研究从剑麻转录组数据中选取8个内参基因[肌动蛋白(ACT2和ACT54)、3-磷酸甘油醛脱氢酶(GAPDH)、亲环蛋白(CYP)、β微管蛋白(TUB)、苹果酸酶(MADH)、转录延伸因子(EF1α和EF1β)],利用GeNorm[17]、BestKeeper[18]和NormFinder[19]统计软件,筛选热麻1号在烟草疫霉侵染、茉莉酸甲酯(MeJA)、水杨酸(SA)和脱落酸(ABA)处理过程中稳定表达的内参基因。本研究不仅改善了剑麻内参基因少的现状,还为剑麻基因表达研究提供了可选择的内参基因,保证了实验结果的真实可靠。
1 材料与方法
1.1 材料
用于烟草疫霉接种的为两年生热麻1号盆栽苗,用于MeJA、SA和ABA处理的为8~10 cm的热麻1号组培苗。菌株为本实验室前期分离保存的烟草疫霉(Phytophthora nicotianae Breda de Haan)。
1.2 方法
1.2.1 病原接种 将保存于PDA斜面的烟草疫霉转接到V8培养基上,28 ℃培养1周后接种。接种前先用酒精棉反复擦拭叶片,然后用灭菌大头针刺伤叶片表面,取直径0.5 cm大小的菌饼将带菌丝面贴于伤口处,用棉花保湿,每隔2 h加1次水,每个生物学重复当作1个处理,每个处理重复3次。分别在接种0、24、36、48、72 h取热麻1号叶片,经液氮速冻后?80 ℃保存备用。
1.2.2 激素处理 取8~10 cm的剑麻组培苗,分别在培养基中加入1 mL SA(10 mmol/L)、MeJA(1 mmol/L)和ABA(0.1 mmol/L)溶液,蓋上盖子,静置放于桌面上;每瓶苗当做1个处理,每个处理重复3次;分别在处理前(0 h)、处理3、6、12、24 h取组培苗叶片,液氮速冻后?80 ℃保存备用。
1.2.3 RNA提取和cDNA合成 用植物RNA提取试剂盒(北京华越洋生物科技有限公司)提取总RNA,取0.5 ?g总RNA用于cDNA第1链的合成。cDNA第1链的合成采用北京全式金生物技术有限公司的TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix反转录试剂盒进行,合成的cDNA按1∶5比例稀释后?20 ℃保存备用。所有操作均按照试剂盒说明书进行。
1.2.4 引物合成 根据转录组测序获得的核酸序列,采用在线引物设计软件Primer Premier3.0[20]设计内参基因引物,引物长度为18~23 bp,Tm值在57~62 ℃之间,产物大小在100~200 bp之间,并在NCBI上对引物的特异性进行检测。引物由上海生工生物工程有限公司合成。引物序列见表1。
1.2.5 内参基因目的片段的PCR扩增 PCR反应体系:2×NOVA Taq-Plus PCR Forest Mix(江苏愚公生命科技有限公司),10 ?L;引物(10 ?mol/L)F 0.2 ?L,R 0.2 ?L;cDNA 2 ?L;灭菌ddH2O 7.6 ?L。PCR扩增程序:先预变性95 ℃,5 min;然后进入35个循环(95 ℃ 30 s,58 ℃ 1 min,72 ℃ 30 s);最后72 ℃,10 min,扩增产物用2.0%琼脂糖凝胶电泳进行检测为单一条带的引物,将进行下一步的荧光定量PCR扩增。
1.2.6 内参基因荧光定量PCR扩增 荧光定量PCR扩增采用北京全式金生物技术有限公司的TransStart? Top Green qPCR SuperMix试剂盒,具体操作按说明书进行。荧光定量PCR反应体系:2×TransStart? Top Green qPCR SuperMix 10 ?L;cDNA 2 ?L;引物(10 ?mol/L)F 0.4 ?L,R 0.4 ?L;灭菌ddH2O 7.2 ?L。扩增程序:先预变性95 ℃,1 min,然后进入45个循环(95 ℃ 10 s,62 ℃ 30 s,72 ℃ 30 s);每个样品设置3次重复,反应结束后进行溶解曲线分析,根据熔解曲线确定产物的特异性。
1.3 数据处理
实时荧光定量RT-PCR后,仪器会自动得出Ct值,首先利用GeNorm[17]、NormFinder[19]和BestKeeper[18]软件分别计算出不同处理下内参基因的表达稳定性(M值)、稳定值(SV值)和变异系数(CV)/标准偏差(SD)值,并根据M值、SV值和CV/SD值按从小到大对内参基因表达稳定性进行排序,值越小对应的内参基因越稳定。最后根据geNorm、NormFinder和BestKeeper软件的排序结果,计算出每个内参基因稳定性综合排序。
2 结果与分析
2.1 内参基因的选择
在转录组数据分析中,基因表达量的计算常用的是FPKM(fragments per kb per million reads)算法[21],其中FPKM值的高低代表基因转录本数的高低。由于剑麻已知的内参基因较少,因此在选择基因时,根据剑麻转录组和表达谱数据,选择转录本数较高的,且在烟草疫霉接种过程中表达稳定的基因作为候选内参基因(表1)。包括肌动蛋白2(ACT2)、3-磷酸甘油醛脱氢酶(GAPDH)、转录延伸因子1-(EF1)、亲环蛋白(CYP)、β微管蛋白(TUB)、苹果酸酶(MADH)、转录延伸因子1-β(EF1β)、肌动蛋白54(ACT54)。
2.2 引物特异性分析
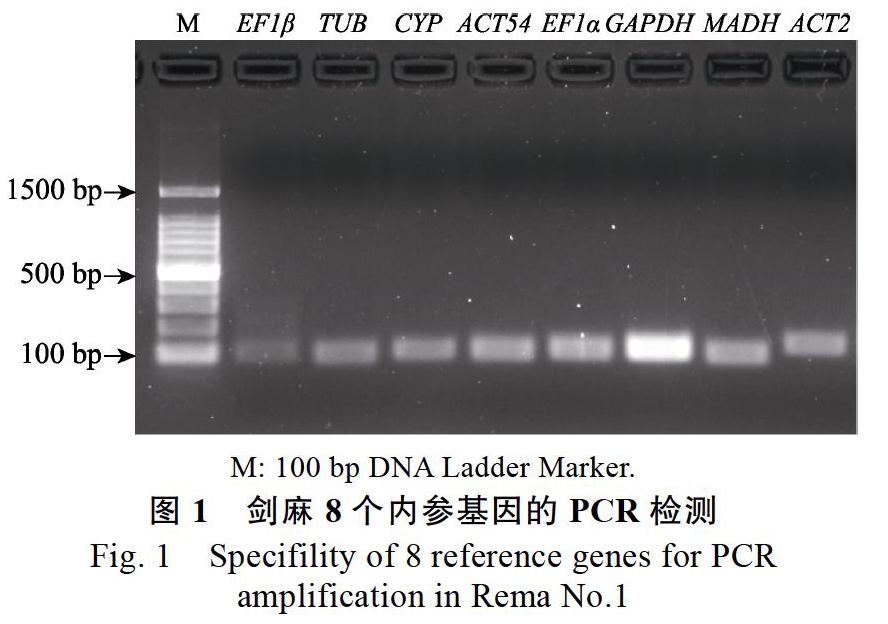
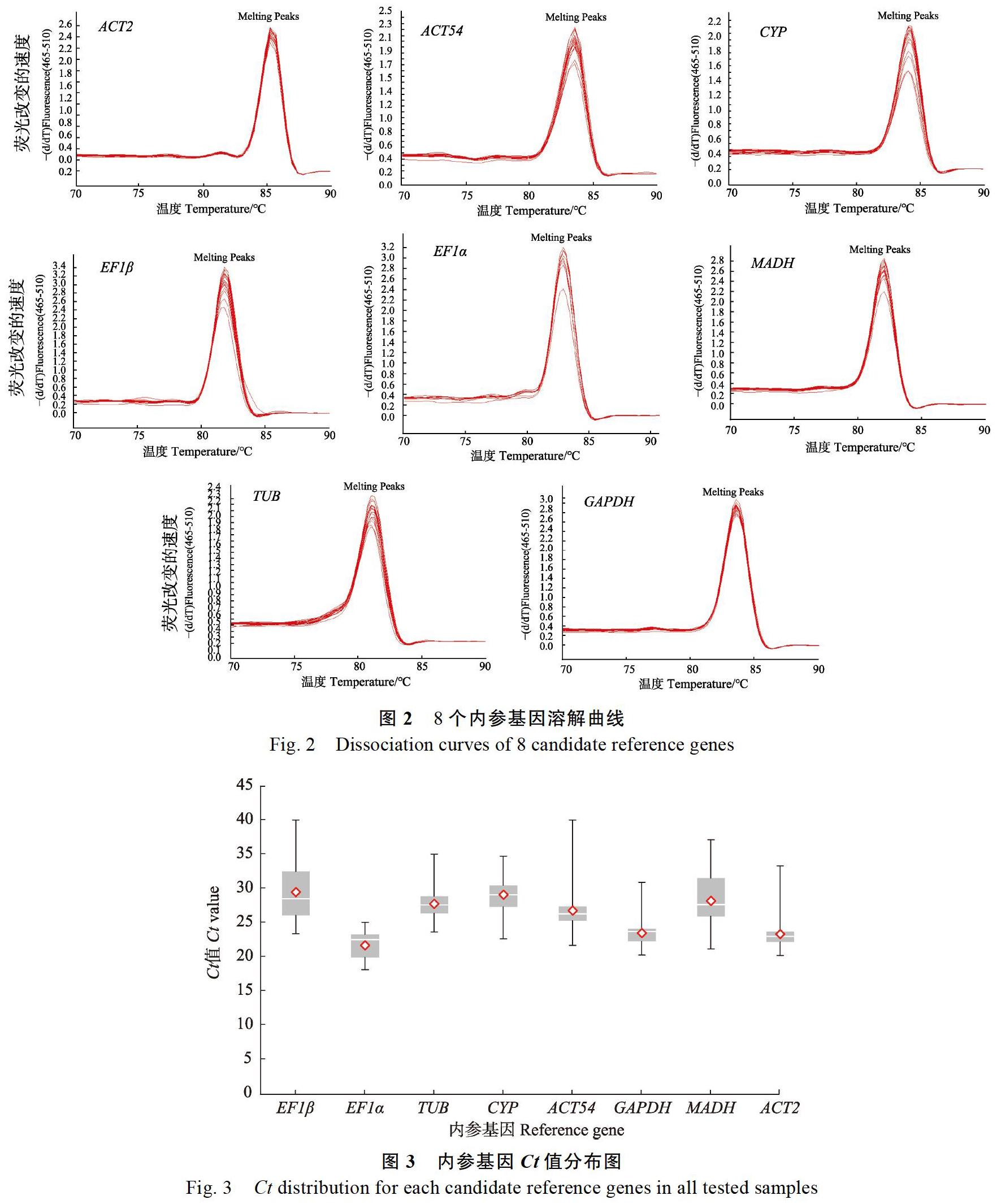
电泳检测结果显示8个候选内参基因均扩增出单一条带,且条带大小与预期产物相符(图1),表明引物特异性较好,可用于实时荧光定量RT-PCR分析。而实时荧光定量RT-PCR熔解曲线分析则表明,8个基因的溶解曲线均呈现显著的单一峰(图2),可用于内参基因表达稳定性分析。
2.3 内参基因表达分析
内参基因的Ct值反映基因的表达水平,Ct值越低表示基因的表达丰度越高。并且同一基因在不同条件下的Ct值变化反应基因表达水平的变化,Ct变化越小则该基因稳定性越好。8个候选内参基因Ct平均值在21.70~29.47之间,其中EF1β的平均Ct值最大,为29.47,即该基因的表达丰度最低,EF1α的Ct平均值最小,为21.70,即该基因的表达丰度最高。从Ct值的分布图可以看出,8个内参基因的表达丰度从高到低依次为EF1α>ACT2> GAPDH>ACT54>TUB>MADH>CYP>EF1β(图3)。
2.4 内参基因稳定性分析
2.4.1 GeNorm分析 GeNorm内参分析软件根据计算出的候选内参基因在不同样品中的表达稳定性来确定最稳定的内参基因,M值越大,则基因的稳定性越低,M值越小,基因的稳定性越高。一般地,M≤1.5才视为稳定表达[17]。GeNorm分析结果表明,在烟草疫霉侵染过程中,EF1α和MADH的稳定值最低,为0.65,TUB的稳定值最高,为1.14,即在烟草疫霉侵染过程中,EF1α和MADH表达最稳定,TUB表达最不稳定。SA处理过程中,EF1α和GAPDH的M值最小,为0.29,ACT2的M值最大,为1.28,即在SA处理过程中,EF1α和GAPDH表达最稳定,ACT2表达最不稳定。MeJA处理过程中,EF1α和ACT2的M值最小,为0.16,MADH的M值最大,为1.96,即在MeJA处理过程中EF1α和ACT2表達最稳定,MADH表达最不稳定。ABA处理过程中,GAPDH和ACT2的M值最低,为0.82,EF1β的M值最高,为3.28,即在ABA处理过程中,GAPDH和ACT2表达最稳定,EF1β表达最不稳定(图4)。
2.4.2 NormFinder分析 NormFinder是结合组内方差与组间方差计算候选内参基因的稳定值进行评价,SV值越小,内参基因表达越稳定[19]。NormFinder分析显示,在烟草疫霉侵染过程中,EF1α表达最稳定(SV=0.30),TUB表达最不稳定(SV=0.78),在SA处理过程中,GAPDH表达最稳定(SV=0.08),ACT2表达最不稳定(SV= 1.79)。在MeJA处理过程中,ACT2表达最稳定(SV=0.06),MADH表达最不稳定(SV=1.86)。在ABA处理过程中,GAPDH表达最稳定(SV= 0.28),ACT2表达最不稳定(SV=3.78)(图5)。
2.4.3 Bestkeeper分析 Bestkeeper是通过计算变异系数和标准偏差来反映内参基因的稳定性,其中变异系数和标准差越小,内参基因的稳定性越好,反之稳定性越差[18]。BestKeeper分析表明,在烟草疫霉侵染过程中,EF1α的CV和SD最小(CV=0.95;SD=0.19),表达最稳定,EF1β表达最不稳定(CV=3.56;SD=0.9)。在SA处理过程中,EF1α表达最稳定(CV=1.3;SD=0.3),ACT2表达最不稳定(CV=5.56;SD=1.34)。在MeJA处理过程中,EF1α表达最稳定(CV=2.39;SD=0.56),MADH表达最不稳定(CV=8.54;SD=2.51)。在ABA处理过程中,EF1α(SD最小,为1.35)和CYP表达最稳定(CV最小,为6.04),ACT54最不稳定(CV=3.46;SD=14.74)(图6)。
2.4.4 综合性分析 对GeNorm、NormFinder和BestKeeper软件分析得到的稳定性排名求几何平均值,得到综合指数排名,指数越小,说明内参基因表达越稳定。由图7可见,8个候选内参基因在烟草疫霉侵染过程中基因表达稳定性由高到低依次为EF1α、MADH、ACT2、GAPDH、TUB、ACT54、CYP、EF1β;MeJA处理过程中基因表达稳定性由高到低依次为EF1α、ACT2、GAPDH、ACT54、CYP、TUB、EF1β、MADH;SA处理过程中基因表达稳定性由高到低依次为EF1α、GAPDH、ACT54、MADH、EF1β、TUB、CYP、ACT2;ABA处理过程中基因表达稳定性由高到低依次为GAPDH、CYP、ACT2、EF1α、TUB、MADH、EF1β、ACT54(图7)。
3 讨论
一般认为,理想的内参基因在不同发育阶段、不同组织以及不同逆境条件下表达稳定且表达水平与目标基因相近,并且常常选择管家基因如ACTIN、TUB、GAPDH、EF1α等作为内参基因[9, 16, 22-23]。但越来越多的研究表明,管家基因的表达水平会受不同实验条件影响而发生变化[24-25],因此,根据具体的实验条件选择稳定的内参基因对基因表达分析尤为重要,并且在多种植物中均有报道[25-27]。
本研究发现,同一基因在不同实验条件下稳定性存在较大差异,如ACT2在MeJA处理过程中表达最稳定,在SA处理过程中表达最不稳定。研究还发现同一基因家族中的不同成员,表达稳定性也存在较大差异。如EF1α和EF1β在烟草疫霉侵染过程中表达最稳定,EF1β最不稳定。Rudu?等[27]也发现野生燕麦中的GAPDH1和GAPDH2的稳定性也存在很大差异。由此可见,内参基因并不是通用的,其选择要根据不同的实验条件而定,同时也说明筛选和开发新的内参基因的重要性。
本研究利用转录组数据开发剑麻新的内参基因,通过表达分析证实了新开发的基因EF1α在MeJA、SA和ABA处理过程中表达较常用的Actin[11-12]更稳定。随着组学时代的发展,利用RNA-Seq挖掘新的内参基因显得尤为突出也十分重要[28-30]。如Gao等[29]利用转录组序列筛选出条斑紫菜适应于失水和低温等非生物胁迫的内参基因MAP、UBC和FHP,在脱水胁迫下稳定表达的内参基因CGS1和UBC,低温胁迫下稳定表达的内参基因MAP、UBC和CGS1。Xu等[30]发现Fb15和eIF-4a在水稻胚乳发育过程中的表达比ACT、GAPDH等11个常用的管家基因更稳定。鉴于此,本研究对剑麻烟草疫霉、SA、MeJA和ABA处理后的内参基因进行表达分析,筛选稳定表达的内参基因,为剑麻下一步的基因功能研究提供可供选择的内参基因,具有十分重要的意义。
参考文献
[1] Corbin K R, Byrt C S, Bauer S, et al. Prospecting for energy-rich renewable raw materials: Agave leaf case study[J]. PLoS One, 2015, 10(8): e0135382.
[2] Escalante A, López Soto D R, Velázquez Gutiérrez J E, et al. Pulque, a traditional Mexican alcoholic fermented beverage: historical, microbiological, and technical aspects[J]. Frontiers in Microbiology, 2016, 7: 1026.
[3] Pereira G M, Ribeiro M G, da Silva B P, et al. Structural characterization of a new steroidal saponin from Agave angustifolia var. Marginata and a preliminary investigation of its in vivo antiulcerogenic activity and in vitro membrane permeability property[J]. Bioorganic & Medicinal Chemistry Letters, 2017, 27(18): 4345-4349.
[4] López-Romero J C, Ayala-Zavala J F, González-Aquilar G A, et al. Biological activities of agave by-products and their possible applications in foods and pharmaceuticals[J]. Journal of the Science of Food and Agriculture, 2018, 98(7): 2461-2474.
[5] Mielenz J R, Rodriguez M, Thompson O A, et al. Development of agave as a dedicated biomass source: production of biofuels from whole plants[J]. Biotechnology for Biofuels, 2015, 8(1): 79.
[6] Stewart J R. Agave as a model CAM crop system for a warming and drying world[J]. Frontier in Plant Science, 2015, 6: 684.
[7] 張燕梅, 赵艳龙, 李俊峰, 等. 剑麻与烟草疫霉互作过程中的转录组研究[J]. 热带作物学报, 2018, 39(3): 540-546.
[8] Wang M L, Medrano J F. Real-time PCR for mRNA quantitation[J]. Biotechniques, 2005, 39(1): 75-85.
[9] Kozera B, Rapacz M. Reference genes in real-time PCR[J]. Journal of Applied Genetics, 2013, 54(4): 391-406.
[10] Zhou W Z, Zhang Y M, Lu J Y, et al. Construction and evaluation of normalized cDNA libraries enriched with full-length sequences for rapid discovery of new genes from sisal (Agave sisalana Perr.) different developmental stages[J]. International Journal of Molecular Sciences, 2012, 13(12): 13150-13168.
[11] Abraham Juárez M J, Hernández Cárdenas R, Santoyo Villa J N, et al. Functionally different PIN proteins control auxin flux during bulbil development in Agave tequilana[J]. Journal of Experimental Botany, 2015, 66(13): 3893-3905.
[12] Gao J M, Yang F, Zhang S Q, et al. Expression of a hevein-like gene in transgenic Agave hybrid No.11648 enhances tolerance against zebra stripe disease[J]. Plant Cell Tissue and Organ Culture, 2014, 119(3): 579-585.
[13] Sandoval S C D, Juárez M J A, Simpson J. Agave tequilana MADS genes show novel expression patterns in meristems, developing bulbils and floral organs[J]. Sexual Plant Reproduction, 2012, 25(1): 11-26.
[14] Wang P H, Xiong A S, Gao Z H, et al. Selection of suitable reference genes for RT-qPCR normalization under abiotic stresses and hormone stimulation in persimmon (Diospyros kaki Thunb)[J]. PLoS One, 2016, 11(8): e0160885.
[15] Thellin O, Zorzi W, Lakaye B, et al. Housekeeping genes as internal standards: use and limits[J]. Journal of Biotechnology, 1999, 75(2-3): 291-295.
[16] Lin F, Jiang L, Liu Y, et al. Genome-wide identification of housekeeping genes in maize[J]. Plant Molecular Biology, 2014, 86(4-5): 543-554.
[17] Vandesompele J, DePreter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes[J]. Genome Biology, 2002, 3(7): RESEARCH0034.
[18] Andersen C L, Jensen J L, ?rntoft T F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer datasets[J]. Cancer Research, 2004, 64(15): 5245-5250.
[19] Pfaffl M W, Tichopad A, Prgomet C, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-excel-basedtool using pair-wise correlations[J]. Biotechnology Letters, 2004, 26(6): 509-515.
[20] Untergasser A, Cutcutache I, Koressaar T, et al. Primer3- new capabilities and interfaces[J]. Nucleic Acids Research, 2012, 40(15): e115.
[21] Mortazavi A, Williams B A, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq[J]. Nature Methods, 2008, 5(7): 621-628.
[22] Dheda K, Huggett J F, Bustin S A, et al. Validation of housekeeping genes for normalizing RNA expression in real-time PCR [J]. Biotechniques, 2004, 37(1): 112-119.
[23] Chervoneva I, Li Y, Schulz S, et al. Selection of optimal reference genes for normalization in quantitative RT-PCR[J]. BMC Bioinformatics, 2010, 11(1): 253.
[24] Sudhakar Reddy P, Srinivas Reddy D, Sivasakthi K, et al. Evaluation of Sorghum Sorghum bicolor (L.) reference genes in various tissues and under abiotic stress conditions for quantitative Real-time PCR data normalization[J]. Frontiers in Plant Science, 2016, 7: 529.
[25] Xu H, Bao J D, Dai J S, et al. Genome-Wide identification of new reference genes for qRT-PCR normalization under high temperature stress in rice endosperm[J]. PLoS One, 2015, 10(11): e0142015.
[26] Narsai R, Ivanova A, Ng S, et al. Defining reference genes in Oryza sativa using organ, development, biotic and abiotic transcriptome datasets[J]. BMC Plant Biology, 2010, 10(1): 56.
[27] Rudu? I, Kepczyński J. Reference gene selection for molecular studies of dormancy in wild oat (Avena fatua L) caryopses by RT-qPCR method[J]. PLoS One, 2018, 13(2): e0192343.
[28] Hu X, Zhang L, Nan S, et al. Selection and validation of reference genes for quantitative real-time PCR in Artemisia sphaerocephala based on transcriptome sequence data[J]. Gene, 2018, 657: 39-49.
[29] Gao D, Kong F, Sun P, et al. Transcriptome-wide identification of optimal reference genes for expression analysis of Pyropia yezoensis responses to abiotic stress[J]. BMC Genomics, 2018, 19(1): 251.
[30] Xu H, Bao J D, Dai J S, et al. Genome-wide identification of new reference genes for qRT-PCR normalization under high temperature stress in rice endosperm[J]. PLoS One, 2015, 10(11): e0142015.
