
利用CRISPR/Cas9系统敲除大肠杆菌tnaA基因
2019-12-19 01:51何京桦乐科易
生物学杂志 2019年6期
何京桦, 乐科易
(复旦大学 生命科学学院, 上海 200433)
L-色氨酸在人和动物体内非常重要,同时在医药、食品和饲料添加剂等方面具有广泛的用途[1-4]。大肠杆菌生长迅速、遗传背景清晰,被认为是生产L-色氨酸的主要菌株。大肠杆菌的tnaA基因编码色氨酸酶(tryptophanase),该酶在正常情况下可以将L-色氨酸降解成丙酮酸、吲哚和氨,从而使碳源进入TCA循环,阻碍L-色氨酸的积累[5-6]。tnaA基因的失活有利于大肠杆菌积累更多的L-色氨酸[7]。
成簇、规则间隔的短回文重复序列(clustered, regularly interspaced, short palindromic repeat,CRISPR)是细菌在长期进化过程中产生的抵抗外源基因入侵的适应性免疫系统[8-11]。CRISPR-Cas9系统为RNA介导的基因组编辑提供了强大的工具,并已广泛应用于包括原核生物和真核生物在内的多种生物中[12-14]。在该系统中,CRISPR RNA(crRNA)通过与转录激活crRNA(trans-activating RNA,tracrRNA)结合形成tracrRNA/crRNA复合物,引导Cas9核酸内酶在目的DNA序列生成双链断裂(double-strand breaks,DSBs)[15-16]。gRNA与靶DNA序列中长约20 bp的片段互补配对,靶序列末端须含有PAM序列(5′-NGG)[17]。
本研究以E.coliw3110-Δ为出发菌株,利用CRISPR/Cas9基因编辑技术对其tnaA基因进行敲除,筛选出tnaA基因缺失菌株,减少色氨酸的降解,从而为L-色氨酸高产菌株的构建奠定基础。
1 材料与方法
1.1 菌株与质粒
E.coliw3110-Δ由本实验室保存,特征是酪氨酸缺陷型,适合于芳香族氨基酸发酵。pCas和pTargetF质粒通过Add gene载体库(http://www.addgene.org/crispr-cas)获得,编号分别为62225和62226。pTargetF-tnaA质粒由本研究构建。
1.2 主要试剂
DNA产物纯化试剂盒(TIAN quick Mini Purification Kit)购于天根生化科技(北京)有限公司。DNA聚合酶(Prime STAR®Max DNA Polymerase)和限制性内切酶DpnI均购于宝生物工程(大连)有限公司。Gibson组装克隆试剂盒(Gibson Assemly Cloning Kit)购自NEB(北京)有限公司。
1.3 gRNA质粒的构建
通过http://www.rgenome.net/cas-offinder/设计tnaA基因的gRNA。将设计出的20 bp序列加入引物gRNAR中。用一步等温Gibson组装法利用Prime STAR®Max DNA 聚合酶将pTargetF、上下游引物gRNA F、gRNA R进行组装[18]。将获得的产物用DpnI酶切,以去除多余的pTargetF。获得的产物转化到E.coliDH5α感受态细胞中,小提质粒,用测序引物gRNAP F/gRNAP R测序鉴定。将构建好的质粒命名为pTargetF-tnaA。本研究所用引物和gRNA寡核苷酸序列见表1。

表1 gRNA和引物
1.4 tnaA基因同源修复供体 DNA的构建
以E.coliw3110-Δ基因组DNA为模板,tnaA L F、tnaA L R为引物,PCR扩增获得片段1,以tnaA R F、tnaA R R为引物,PCR扩增获得片段2。以AmlF和AmlR为引物,将片段1和片段2用重叠延伸PCR法获得同源修复供体DNA。
1.5 感受态细胞的制备及细菌的转化
将pCas质粒转化到E.coliw3110-Δ感受态细胞中,转化产物涂布到含有卡那霉素(50 μg/mL)的LB琼脂上(30 ℃培养),挑选获得含有pCas质粒的E.coliw3110-Δ。将含有pCas质粒的E.coliw3110-Δ按照文献[14]描述的方法制备感受态细胞。当感受态细胞生长至其OD600为0.1时,加入终浓度为10 mmol/L的阿拉伯糖,以诱导λ-Red介导的同源重组[19]。当OD600=0.6时收集菌体,将100 ng gRNA质粒pTargetF-tnaA和500 ng供体DNA加入到约100 μL含pCas质粒的E.coliw3110-Δ感受态细胞中,进行电转(2 mm cuvette, 2.5 kV)。电转后立即在1 mL LB液体培养基中悬浮细胞,30 ℃复苏1 h,涂布至含有卡那霉素(50 μg/mL)和壮观霉素(50 μg/mL)的LB琼脂培养基上。30 ℃过夜培养,用引物tnaAP F和tnaAP R对转化子进行PCR检测,并进行测序鉴定,将测序结果和原基因序列进行对比,检测tnaA基因是否敲除成功。
1.6 pTargetF-tnaA和pCas的消除
将鉴定正确的含有pCas和pTargetF-tnaA的克隆接种于含有卡那霉素(50 μg/mL)和IPTG(异丙基-β-D-硫代半乳糖苷,0.5 mmol/L)的2 mL LB培养基中,培养8到16 h后涂布到含有卡那霉素(50 μg/mL)的LB琼脂上。然后以克隆对壮观霉素(50 μg/mL)的敏感性确认pTargetF-tnaA已经被消除。将克隆在43 ℃培养过夜以消除pCas质粒[20]。
1.7 敲除效率的优化及检测
为提高该敲除系统对大肠杆菌tnaA基因的敲除效率,在100 μL感受态细胞中,将pTargetF-tnaA质粒加入量由普遍采用的100 ng提高到300 ng, 供体DNA片段加入量由普遍采用的400 ng提高到1.5 μg进行转化,用引物tnaAP F和tnaAP R对转化子进行PCR鉴定和测序鉴定,计算该系统对tnaA的敲除效率。
2 结果与分析
2.1 gRNA的构建
用一步等温Gibson组装法利用Prime STAR®Max DNA 聚合酶将pTargetF质粒、上下游引物进行组装。获得的产物转化到E.coliDH5α感受态细胞中,小提质粒,将提取的质粒pTargetF-tnaA送检测序,测序结果正确,可用于下一步试验。
2.2 tnaA基因同源修复供体 DNA的构建
分别以tnaA L F/R 和tnaARF/R为引物,PCR扩增获得了片段1和片段2,以AmlF和AmlR为引物,利用片段1和片段2用重叠延伸PCR法获得了同源修复供体 DNA。
2.3 tnaA基因的敲除及敲除效率的检测
pCas质粒成功转入E.coliw3110-Δ中。pTargetF-tnaA质粒和供体DNA成功共转入含有pCas质粒的E.coliw3110-Δ中。用横跨tnaA基因同源修复供体DNA的鉴定引物tnaAP F和tnaAP R扩增目的条带来鉴定tnaA基因的敲除情况。结果(图1)显示,tnaA基因没被敲除修复的菌,PCR显示为1150 bp的条带,反之出现750 bp的目的条带。测序结果与设计的同源臂序列是一致的,表明敲除后的tnaA基因在人工设计的供体序列下发生了同源修复。挑取12个克隆进行PCR鉴定和测序鉴定,9个克隆敲除成功,该敲除系统对大肠杆菌tnaA的敲除效率为75%。以上结果均表明,本研究构建的CRISPR/Cas9系统在人工同源修复下,可对大肠杆菌tnaA基因进行高效敲除,图2是敲除模式示意图。
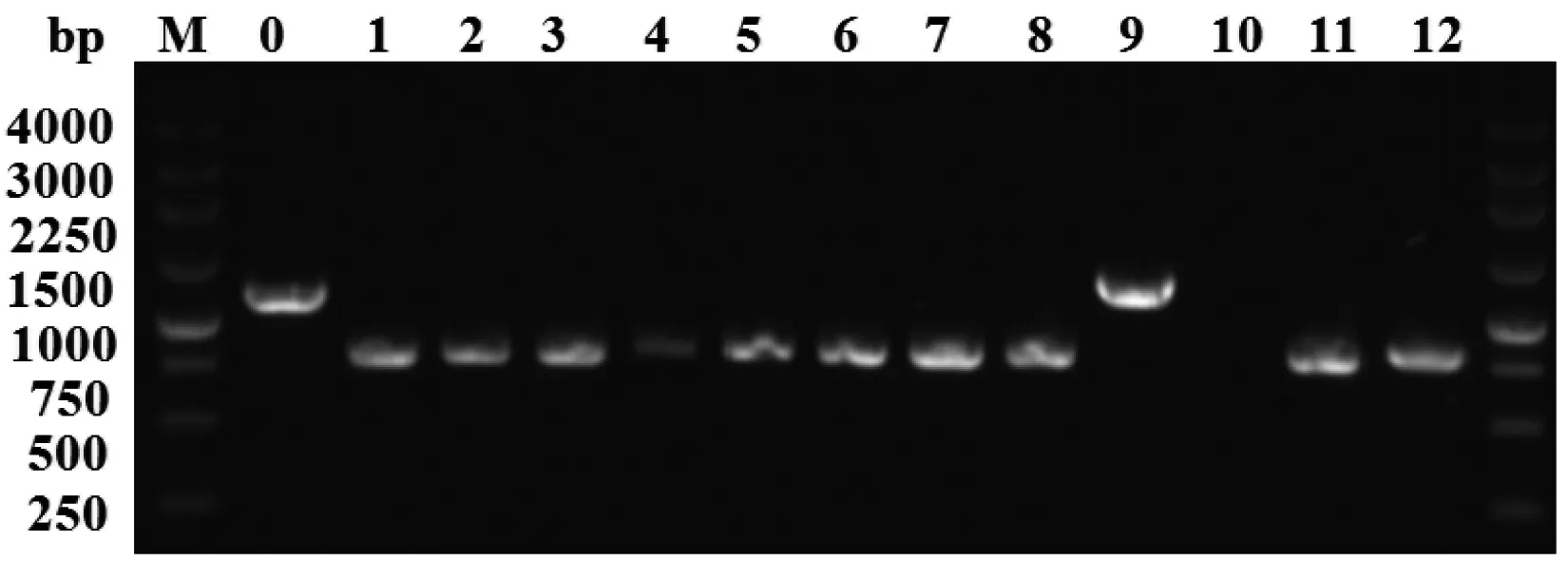
M:相对分子质量;0:原始菌的PCR产物;泳道4、9、10:未敲除成功的PCR产物,大小约为1150 bp;泳道1、2、3、5、6、7、8、11和12:敲除tnaA后的PCR产物,大小约为750 bp
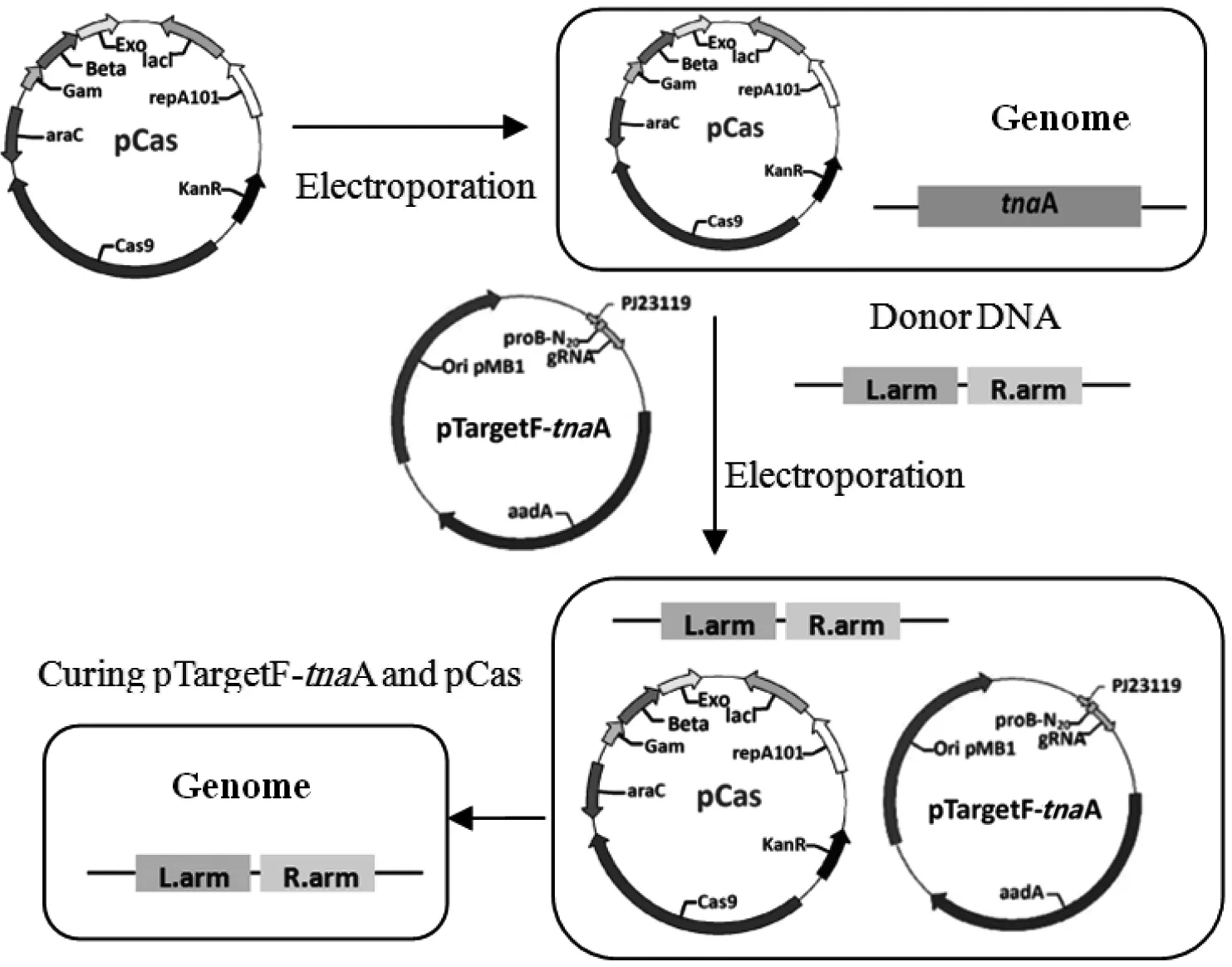
图2 利用CRISPR/Cas9系统进行基因敲除示意图
3 讨论与结论
CRISPR-Cas9是细菌以及古细菌在长期进化过程中形成的一种适应性免疫机制,用以对抗入侵的病毒及外源DNA。CRISPR-Cas9基因编辑技术,则是对目的基因进行特定的DNA修饰的技术,该技术已应用于细菌、真菌、斑马鱼以及人类细胞的基因编辑。
在该技术中,一般是将CRISPR-Cas9技术与同源重组技术结合起来,构建含Cas9、sgRNA和Donor序列的重组载体,对目标基因进行有效准确的编辑。当然该技术也在不断地被改进优化, Jiang等人在Cas9质粒构建时引入了Red重组系统,供体DNA序列不需要克隆到载体上,而是直接与质粒共转化入宿主菌,使得该技术可以对多个基因同时进行高效的敲除[14]。
为了避免双链断裂DNA被同源修复后,gRNA/Cas9复合体对其重复切割,Wang等人采用了两步法策略,第一步使用CRISPR/Cas9系统用20 bp人工序列替换了原始间隔序列,以避免gRNA/Cas9复合体的重复切割;第二步使用第二个gRNA识别引入的人工序列进行基因突变,同时人工序列被还原为原始序列,从而只引入一个点突变,且不会改变PAM或不会在基因组中插入额外的沉默突变[20]。
本研究在构建质粒时采用了Gibson等温一步拼接法,该方法是目前最常用的新兴克隆方法之一, 能在5′核酸外切酶、DNA聚合酶和DNA连接酶的协同作用下直接组装多个重叠DNA分子,这种组装方法可以无缝地构建基因, 是一种有用的分子工程工具。
本研究成功构建了大肠杆菌tnaA基因CRISPR/Cas9敲除系统,敲除效率75%。该系统将来也可应用于大肠杆菌其他基因的敲除,为构建L-色氨酸高产大肠杆菌提供有效的基因敲除工具。
猜你喜欢
北方牧业(2022年9期)2022-11-22
中国医药导报(2021年35期)2022-01-20
汉字汉语研究(2021年2期)2021-08-30
家畜生态学报(2020年7期)2020-07-14
华南师范大学学报(自然科学版)(2020年3期)2020-07-01
首都医科大学学报(2019年5期)2019-10-24
汉字汉语研究(2019年2期)2019-08-27
中国饲料(2019年5期)2019-01-10
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
