
苯并咪唑类化合物靶向抗肿瘤作用及其机制研究进展
2019-12-19 01:48:12丁孺孺滕梦婷胡佳颖
生物学杂志 2019年6期
丁孺孺, 滕梦婷, 胡佳颖, 张 鹏
(江苏师范大学 生命科学学院, 徐州 221116)
开发新型、高效、低毒的抗癌药物,选择性地杀死肿瘤细胞或抑制肿瘤细胞生长而不影响非癌组织是抗癌药物研发中面临的最大难题[1]。肿瘤分子靶向治疗是根据导致癌症的相关分子(蛋白质分子或DNA片段)专门设计的药物,这些药物可以特异性地识别并杀死肿瘤细胞。与传统化疗药物相比,靶向药物对正常细胞或组织的毒副作用很小,是目前抗肿瘤药物研发的热点之一[2]。
苯并咪唑(Benzimidazole)是一种含有两个氮原子的苯并杂环化合物,最初是因为其为维生素B12的重要组成部分而被关注(见图1)[3]。苯并咪唑类衍生物具有很多生物活性,其中包括杀菌[4-5]、抗病毒[6-7]、消炎[8-9]及镇痛[10-11]等。目前苯并咪唑已经在临床有很多应用,例如奥美拉唑作为质子泵抑制剂治疗十二指肠溃疡和甲苯咪唑作为驱虫剂能与寄生虫肠细胞微管蛋白特异性结合干扰其细胞微管从而达到驱虫的效果。一些苯并咪唑衍生物被证明能在体外及体内选择性地抑制内皮细胞生长和血管生成[12-13]。国内外关于苯并咪唑类衍生物的合成及其抗肿瘤机制的研究越来越多,笔者在数据库可搜到近5年的相关文献146篇,并且每年呈现上升的趋势,由此可以推测以苯并咪唑为骨架合成的衍生物可能是今后抗肿瘤药物研发的热点方向之一。
1 拓扑异构酶抑制剂
DNA拓扑异构酶是细胞核中与DNA转录、修复和染色质组装有关的酶。与正常细胞相比,DNA拓扑异构酶在肿瘤细胞中稳定地高水平表达。通过抑制其活性可以抑制肿瘤的发生和发展。因此,DNA拓扑异构酶成为肿瘤治疗的一个重要靶点[14-15]。
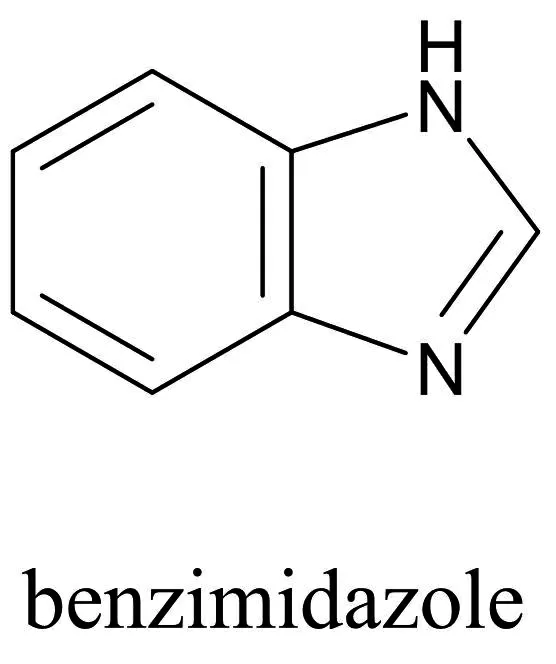
图1 苯并咪唑的化学结构式
Gao等把吖啶和苯并咪唑结合在一起,合成了化合物8a-8q,由于吖啶可以插入DNA并结合到DNA链中相邻的碱基对之间。因此,二者的连接可以增加化合物对DNA的亲和性。 通过8a-8q的细胞增殖实验,发现化合物8l对人慢性髓系白血病细胞K562和人肝癌细胞HepG2的增殖有较好的抑制作用,其IC50分别为2.68 μmol/L和8.11 μmol/L,并且证明其具有结合DNA及抑制拓扑异构酶I的作用。JC-1染色证明化合物8l可以降低K562细胞的线粒体膜电位。Western Blot结果分析表明,随着化合物8l的浓度升高caspase-7,caspase-9,caspase-3和PARP(poly ADP-ribose polymerase)的切割体比例依次升高,说明化合物8l可能是通过线粒体途径诱导K562细胞凋亡[16]。Singh等合成了具有供电子(OCH3)或者吸电子基团(F,Cl)的1位和3位取代的双苯并咪唑,通过实验发现,苯环上引入具有吸电子性质的卤素基团可以增强化合物与ctDNA的亲和度。Singh通过实验证明有吸电子基团(F,Cl)的 6a和6b比引入甲氧基等供电子基团的衍生物显示出对肿瘤细胞更大的毒性[17](图2)。
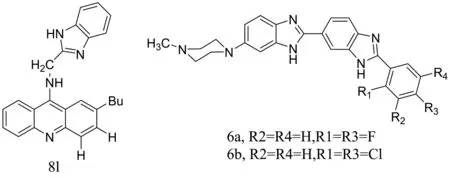
图2化合物8l,6a和6b结构
2 PARP抑制剂
DNA一直处于损伤和修复的动态平衡中,当DNA受到损伤之后,细胞就会启动DNA损伤修复机制,通过转录某些修复蛋白进行DNA修复,使细胞继续存活,如果损伤部位无法修复便会启动凋亡机制使细胞凋亡。PARP(Poly ADP-ribose polymerase)是细胞核内的一种参与DNA检测和修复的酶,当DNA单链受到损伤后,多聚(ADP-核糖)聚合酶和一些其他修复蛋白会被募集到损伤部位进行DNA修复。通过抑制PARP可以导致肿瘤细胞的DNA无法正常修复而加速其死亡。PARP抑制剂最初被用于增强放疗或者化疗的作用,直到2005年研究人员发现,brca1和brca2基因突变的肿瘤患者对PARP抑制剂的敏感度提高了约1000倍,其原因是brca突变使肿瘤细胞的DNA修复能力受限,从而增加了细胞毒性[18]。第一个被FDA批准上市的PARP抑制剂是阿斯利康公司研发的Olaparib,用于治疗brca突变的晚期卵巢癌。
Veliparib(ABT-888)是一种口服的PARP-1和PARP-2抑制剂,其作用是延迟化疗或放疗引起的DNA损伤修复[19]。另外Veliparib可能在DNA损伤部位产生稳定的PARP-1/2复合物,这些复合物的产生增加了细胞的毒性。Donawho等通过实验证明Veliparib可以穿过血脑屏障,并且能在异种移植肿瘤模型中增强替莫唑胺、顺铂、环磷酰胺和放疗的作用[20]。Penning等通过实验证明Veliparib对于PARP-1 和 PARP-2具有很好的抑制的效果(Ki=5 nmol/L),而且对C41细胞的EC50(PARP活性抑制50%的有效浓度)为2 nmol/L[21]。2014年1月15日,雅培生命宣布Veliparib 与化疗药物卡铂联合进行III期临床试验,用于评价其治疗早期三阴性乳腺癌的安全性和有效性(图3)[22]。
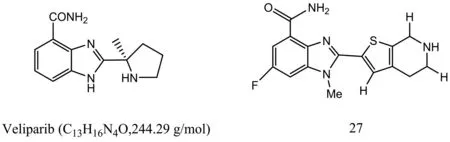
图3 Veliparib和化合物27的化学结构式
Chen课题组设计并合成一系列了4,5,6,7-四氢噻吩吡啶-2-基苯并咪唑羧酸酰胺类化合物,部分化合物能有效地抑制PARP的活性,其中化合物27号对PARP-1 和 PARP-2的IC50分别为18和42 nmol/L。同时,该化合物可以选择性地抑制brca2突变的V-C8细胞增殖,其CC50为920 nmol/L[23]。
3 微管蛋白抑制剂
微管作为细胞骨架在维持细胞的形状,细胞内信号转导和有丝分裂起着重要作用,它的主要组分是α和β 微管蛋白异二聚体[24]。紫杉醇作为常见的微管蛋白抑制剂,1992年被FDA批准上市用于卵巢癌和乳腺癌的治疗并显示出非常好的效果,它可以加速微管蛋白的聚合并抑制其解聚,从而使肿瘤细胞阻滞在G2/M期,最终诱导细胞凋亡[25]。紫杉醇最初来源于濒临灭绝的植物红豆杉,但是,该植物生长缓慢而且紫杉醇含量极少。严重制约了紫杉醇的使用。因此人工合成以微管蛋白为靶点的抗肿瘤药物显得尤为重要。
Ahmed研究组将苯并咪唑和羟吲哚结合起来合成了一批化合物,通过细胞毒性实验发现化合物5c和5p可以有效抑制人乳腺癌细胞MCF-7的增殖,其IC50为1.84 μmol/L和1.97 μmol/L,而且化合物5c和5p可以很好地抑制微管蛋白聚合,IC50分别为1.12 μmol/L和1.59 μmol/L。另外这两个化合物对正常人胚胎肾细胞HEK-293和正常猴肾细胞Vero的毒性很低,显示出很好的选择性。用5c和5p处理MCF-7细胞后进行的流式细胞检测分析发现细胞被阻滞在G2/M期。通过分子对接发现化合物可与微管蛋白相互作用并有效结合(图4)[26]。Lakshma等后来对化合物5c和5p进行了凋亡机制研究,结果表明5c和5p可以通过上调促凋亡蛋白Bax,下调抗凋亡蛋白Bcl-2,并且激活caspase-9来介导人类乳腺癌细胞MCF-7凋亡[27]。Wang合成一批苯并咪唑脲类衍生物,它们对人体肿瘤细胞系的IC50值在纳摩尔范围其中化合物6o的抗肿瘤活性最好。通过免疫荧光和细胞周期实验发现,当6o 的浓度超过100 nmol/L时,人大细胞肺癌细胞NCI-H460细胞纺锤体的形成受到抑制,最终阻滞在G2/M期,计算机模拟显示,β-微管蛋白的Glu71(71位谷氨酸残基)和α-微管蛋白的Asp251(251 位天冬氨酸残基)与化合物6o的苯并咪唑基团形成两个关键氢键,α-微管蛋白的Arg2(2 位精氨酸残基)与化合物6o的脲基形成一个氢键;同时α-微管蛋白的Arg2(2 位精氨酸残基)与6o的氰基能够与形成另一个重要的氢键(图5)[28]。这些研究显示了苯并咪唑类化合物作为微管蛋白抑制剂有着非常好的发展前景,随着研究的不断深入,相信会有更多以微管蛋白为靶点的高效、低毒抗癌化合物应用在临床上。
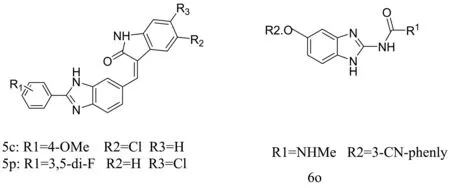
图4 化合物5c,5p和6o的化学结构式
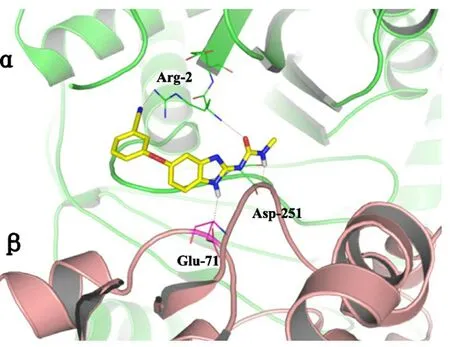
图5 化合物6o分子对接模式图[27]
4 稳定G-四链体DNA及抑制端粒酶活性
人体端粒由富含鸟嘌呤(G)的DNA重复序列组成, 该序列在一定的条件下可以形成G-四链体 DNA 的结构。一些小分子化合物可以诱导该结构的形成并使之稳定, 通过抑制端粒酶的活性降低癌基因的转录表达[29]。
Yadav等分别合成了一系列在C1上有芳环和在C3上带有取代苯并咪唑部分的β-咔啉-苯并咪唑衍生物。通过实验发现5a(3-(6-甲氧基-1H-苯并[d]咪唑-2-基)-1-(4-甲氧基苯基)-9H-吡啶并[3,4-b]吲哚)在低剂量就可以稳定G-四链体DNA并抑制端粒酶活性。实验结果显示HeLa细胞在经化合物5a(3 μmol/L)处理24 h后有17%的细胞发生凋亡,48 h后有50%被阻滞在G1期。分子对接实验结果表明,5a对四链体DNA表现出高的稳定性的原因是由于其-OCH3,-NH基团可以与G-四链体DNA的dG3,dA7之间形成了氢键(图6)[30]。以稳定G-四链体DNA及抑制端粒酶活性为思路来设计合成抗癌药物具有良好的发展前景,但苯并咪唑类化合物在这方面的应用还很少,具有非常大的发展潜力。
5 CK2蛋白激酶抑制剂
蛋白激酶可以通过磷酸化和去磷酸化调节蛋白质的活性,它在细胞信号转导中起着十分重要的作用。通过蛋白质的逐级磷酸化,使信号逐级放大,进而引起细胞反应。蛋白激酶功能失常将导致细胞生长、分化、代谢等功能异常, 最终导致肿瘤的产生[31]。所以,用蛋白激酶为靶点的抗肿瘤药物成了研究的重点,目前已有多个针对蛋白激酶靶点的药物上市。第一个上市的小分子蛋白激酶抑制剂是Imatinib(商品名为 Gleeve),它被称为肿瘤靶向治疗的里程碑[32]。关于苯并咪唑类化合物作为激酶抑制剂的研究也有相关报道。
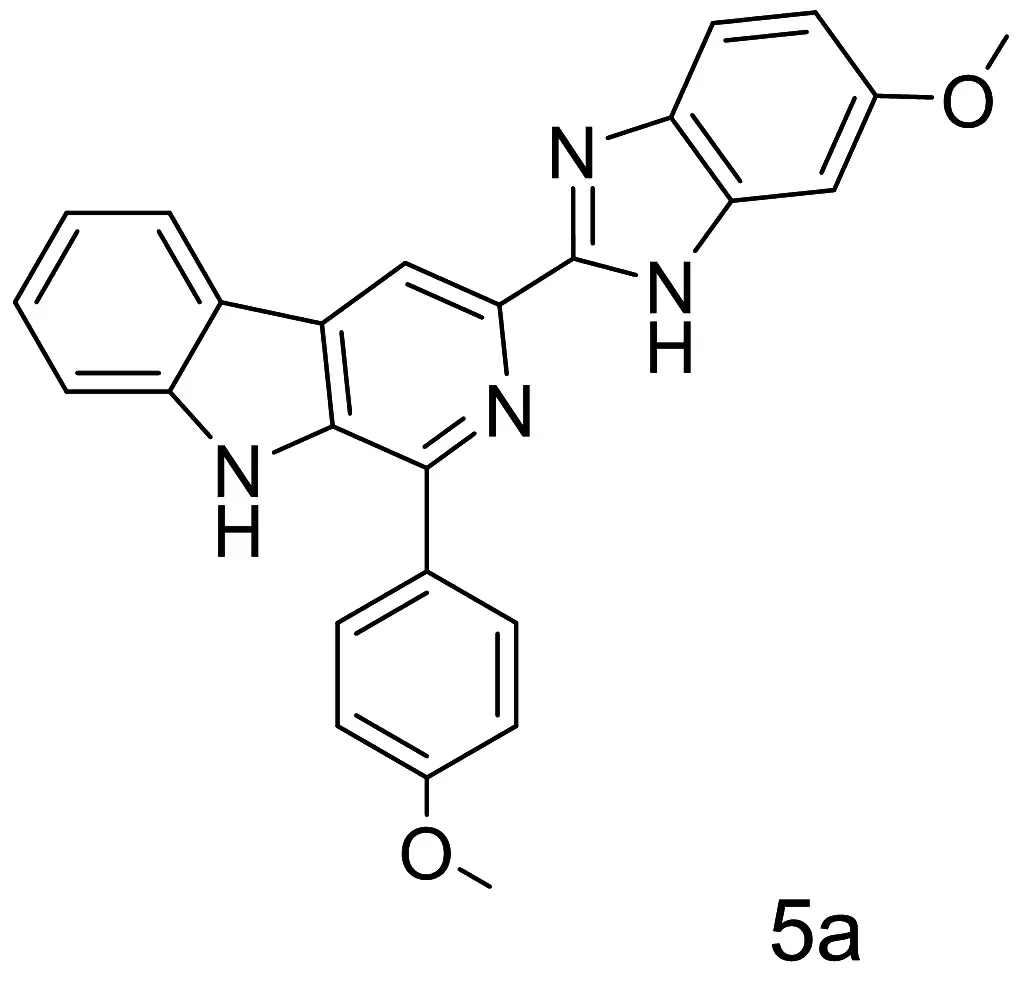
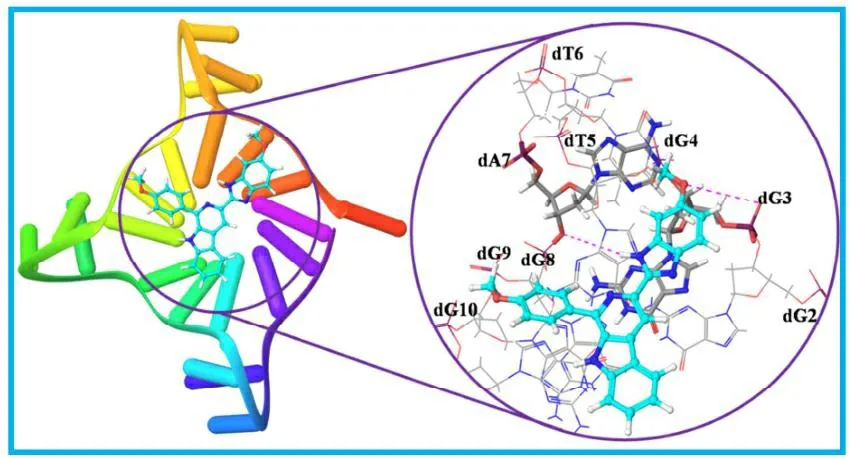
图6化合物5a结构式及其推测结合模式分子图像分析[29]
CK2(Casein kinase Ⅱ)作为多底物蛋白激酶,有300多种作用底物,这些底物对细胞生理功能产生了重要的作用,其中包括细胞代谢和运动、DNA复制和转录、细胞信号的加工与转导、细胞的增殖和凋亡、RNA 加工和翻译[33]。目前CK2抑制剂中研究最多的是4,5,6,7-四溴-1H-苯并三唑(TBBt,IC50为0.32 μmol/L)、4,5,6,7-四溴-1H-苯并咪唑(TBBi,IC50为1.3 μmol/L)及其衍生物(图7)。Chojnacki等根据多数CK2抑制剂都含有羟基或羧基,于是合成了含有羟基或羧基取代的TBBi和TBBt衍生物。通过实验发现被叠氮基取代的TBBi衍生物(3和5)的Ki的范围在2.34~3.28 μmol/L,被羧基取代的三唑环衍生物(7和10)的范围在0.91~1.96 μmol/L。MTT结果显示,TBBi及其衍生物2、3、5、9在50 μmol/L的浓度下能使人急性淋巴细胞白血病T淋巴细胞CCRF-CEM的生长率降到约6%,MCF-7的生长率降到约20%[34]。
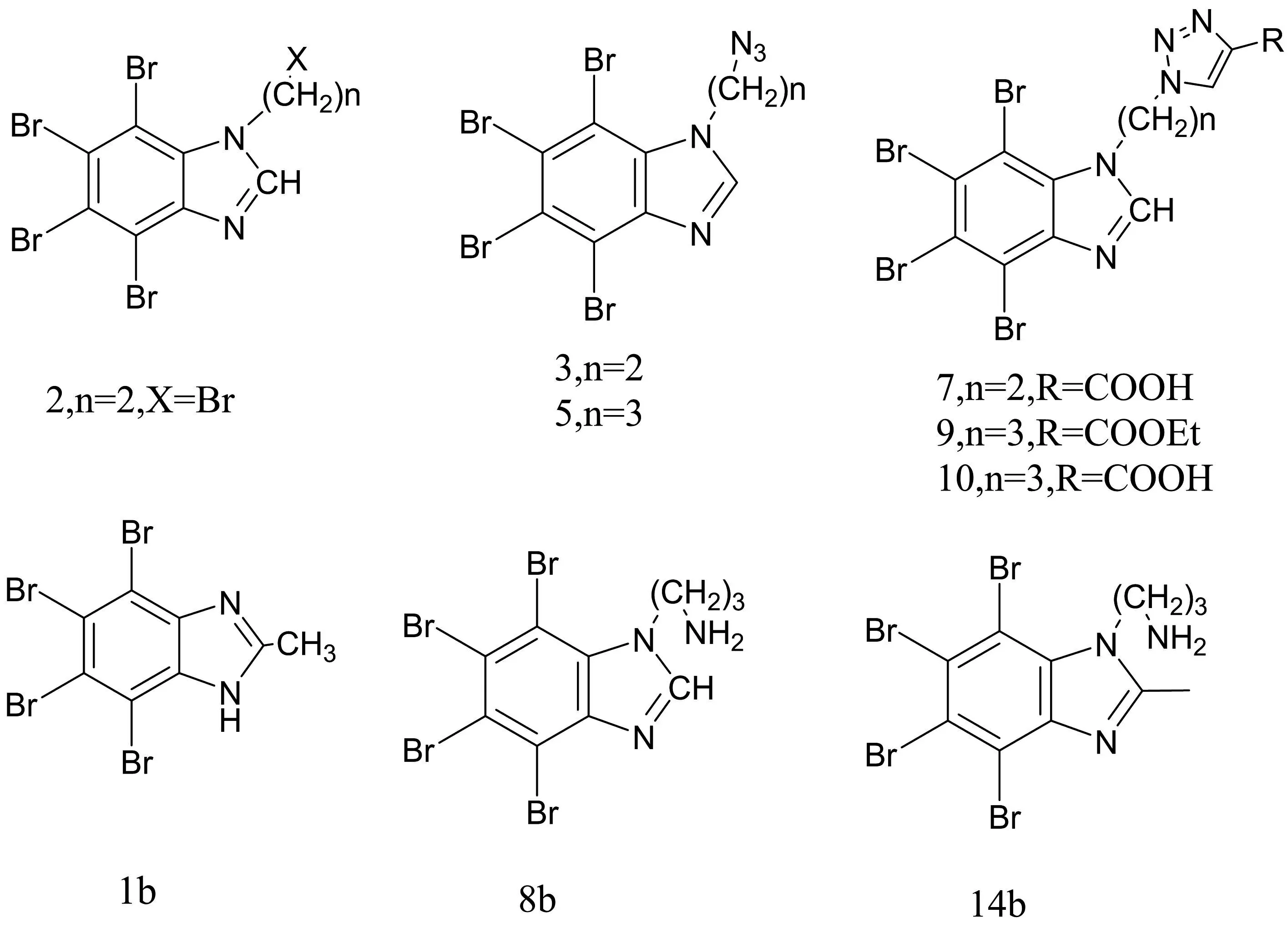
图7 TBBi衍生物的化学结构式
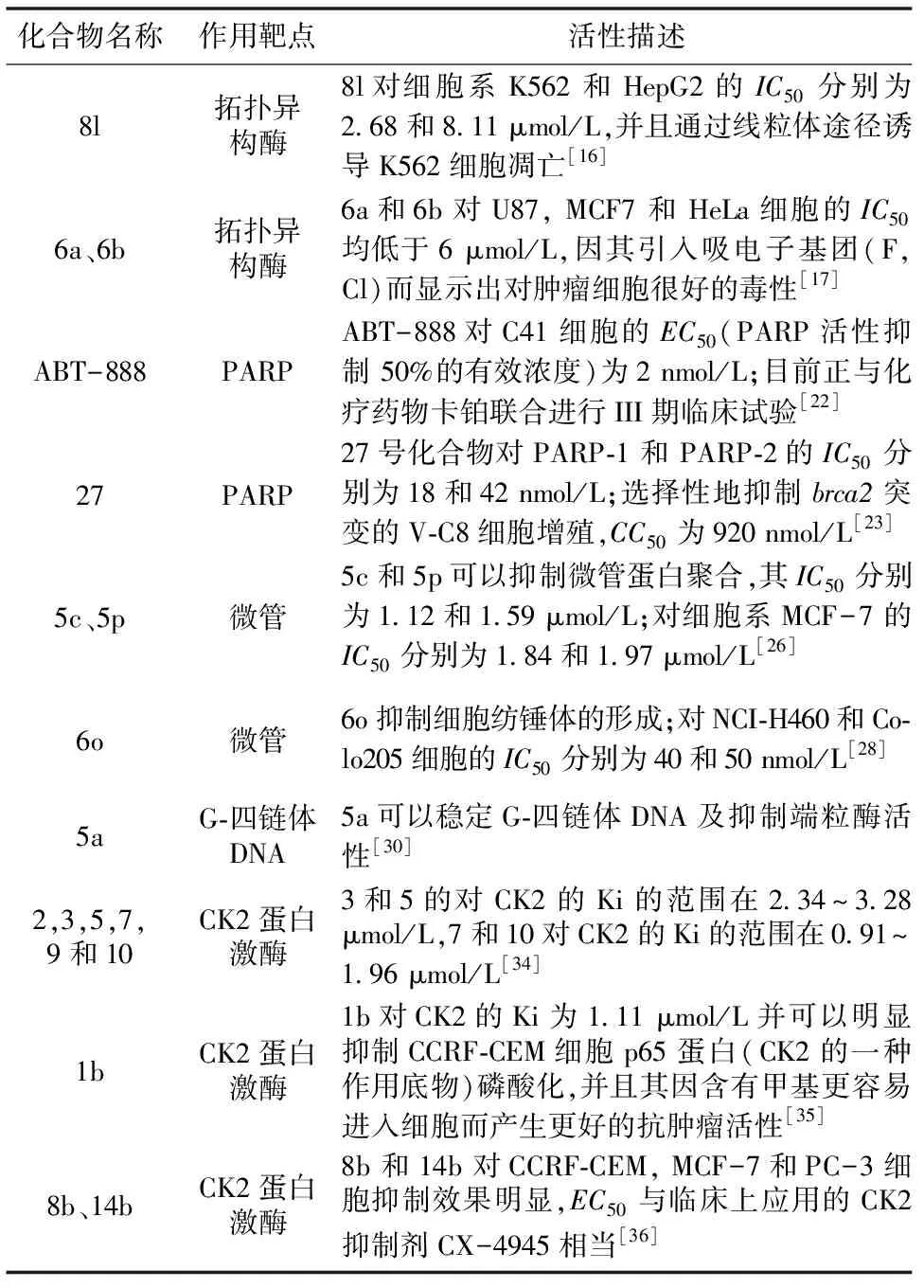
表1 本文所综述化合物汇总
激酶抑制剂选择性低,毒副作用大的问题给此类抗肿瘤药物研发带来巨大挑战[37]。目前文献报道的苯并咪唑类CK2抑制剂虽然对肿瘤细胞显示出较好的作用,但缺乏对正常细胞毒性的测定,限制了对该类化合物抗肿瘤价值的评估,因此对于苯并咪唑类CK2抑制剂的研究工作还需要进一步开展。
6 小结与展望
由于苯并咪唑具有优良的杂环结构,所以在药物研发方面占有一席之地。研究表明,当苯并咪唑被脂族、芳族或杂环部分修饰之后可明显提高其抗肿瘤活性。目前已经发现苯并咪唑类化合物对于拓扑异构酶、端粒酶、微管蛋白、去乙酰化酶、PARP 等肿瘤药物靶点等多方面应用,因此,以苯并咪唑为骨架研发药效更高、毒副作用更小、成药性更强的化合物是今后抗肿瘤药物研发的热点方向之一(表1)。笔者所在课题组根据文献报道设计并合成一系列苯并咪唑类衍生物,并对其抗肿瘤活性做了较为深入的研究,目前已经筛选出几种低剂量下对于人非小细胞肺腺癌细胞A549、乳腺癌细胞MCF-7、三阴性乳腺癌细胞MDA-MB-231等细胞系抑制效果较好的苯并咪唑类衍生物。其中,化合物BZ-18 可以有效地抑制三阴性乳腺癌MDA-MB-231细胞迁移,BZ-25对三阴性乳腺癌MDA-MB-231、HCC1937和BT-483的IC50低于1 μmol/L,远低于人正常细胞(5 μmol/L),表现出较好的选择性。目前,实验室正在开展对化合物BZ-25作用靶点和作用机制的研究。虽然近年来关于苯并咪唑衍生物合成和抗肿瘤作用研究很多,但多数研究者将注意力集中在如何增加其抗肿瘤活性,没有对药物的选择性给予足够的重视。如何针对该类药物的结构特点减小它们对人体正常细胞的毒性将是该类药物分子设计的发展方向。例如,针对发生DNA损伤修复能力发生突变的肿瘤细胞可以设计出效果更好的PARP抑制剂,将会对这类肿瘤产生非常好的选择性杀伤作用。另外由于苯并咪唑类化合物的优良杂环结构,可以针对能够产生协同作用的多条信号通路设计出多靶点化合物,这样将会产生更好的效果和更高的选择性。总之,苯并咪唑类化合物在新药设计方面具有极大的潜力,相信在不久的将来会有更多高效低毒并且价格惠民的治疗癌症的该类新药出现在市场上。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:40
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29 01:21:32
中国资源综合利用(2017年2期)2018-01-22 02:44:58
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
中外医疗(2016年15期)2016-12-01 04:25:49
合成化学(2015年2期)2016-01-17 09:03:42
合成化学(2015年10期)2016-01-17 08:56:26
化工进展(2015年6期)2015-11-13 00:29:04
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
无机化学学报(2014年10期)2014-02-28 17:33:13
