
高氨氮浓度对产甲烷厌氧发酵过程中微生物活性及转录组的影响
2019-12-06 07:00贾传钊李香真肖洪文
中国沼气 2019年4期
贾传钊, 李香真, 肖洪文, 章 淼,
(1.中国核动力研究设计院, 成都 610041; 2.中国科学院成都生物研究所,中国科学院环境与应用微生物重点实验室, 环境微生物四川省重点实验室, 成都 610041)
厌氧发酵可在处理有机废弃物的同时产生甲烷气体[1]。发酵系统中的氨氮主要来自于发酵底物中蛋白质、氨基酸以及尿素等的水解[2],能被微生物直接利用并对发酵系统具有缓冲作用[3]。适宜的氨氮浓度对厌氧发酵系统甲烷产生和稳定性的维持具有促进作用。但高氨氮也会对发酵过程产生不利影响[4],主要是通过对发酵系统中微生物群落结构和活性的影响,从而影响整个反应系统的稳定性。由于产甲烷古菌比水解酸化细菌对氨氮更加敏感[5-6],因此,高氨氮对产甲烷古菌会产生更显著的影响。前期的一些研究揭示了厌氧发酵系统中的微生物群落结构[7],但对微生物活性以及相关的代谢过程研究较少。
转录组信息能够有效帮助研究特定时间与特定环境条件下活性微生物的基因表达水平,准确预测不同环境条件下起作用的微生物[8-10]。并且随着测序技术的不断发展,转录组测序中存在的一些问题逐步得到解决[11]。目前微生物转录组已在动物肠道微生物、土壤、淤泥等方面得到了广泛的应用[12-13]。应用转录组测序不仅能够使我们更好地理解反应系统对外界环境因子冲击的响应,也可利用分析得到的结果对反应系统的环境参数进行调整,使反应系统具有更好的稳定性。
在本研究之前,基于IlluminaMiseq高通量测序技术,已经完成了不同氨氮浓度条件下厌氧发酵系统中微生物群落结构的研究[14]。本研究主要目的是考察高氨氮浓度(>5000 mg·L-1)条件下起作用的活性微生物群落,以及高氨氮对产甲烷古菌的影响。
1 材料与方法
1.1 厌氧反应器的建立与样品采集
厌氧反应器的建立与之前的试验相同[14],采用总固体含量为6%的猪粪作为厌氧发酵底物,在pH值为7.0±0.1,温度为35℃±2℃,水力停留时间为8天的条件下运行。选取起始氨氮浓度为1000 mg·L-1(对照组发酵液中氨氮的初始含量),5500 mg·L-1和7000 mg·L-1的反应器进行研究,并将不同氨氮浓度的反应器分别命名为R1,R2和R3。在发酵第21天采集发酵液重复样品(3个),于12000 rpm离心收集沉淀部分,并冷冻于-80℃以备RNA的提取。
1.2 RNA提取、反转录及16S rRNA基因扩增
将0.3 g固体样品与玻璃珠置于同一个容器中,通过振荡释放样品中的核酸。样品中RNA的提取采用细胞/细菌总RNA提取试剂盒(Cat.No.DP430;TIANGEN,China),按照试剂盒所提供的方法和步骤,使得RNA分离与纯化。得到的RNA采用凝胶电泳和Nanodrop分光光度计进行质量检验(Nanodrop 2000c;Thermo Scientific, USA)。随后,以总RNA为模板,在反转录酶的催化作用下生成cDNA的第一条链,再在DNA聚合酶等作用下生成cDNA的第二条链。这一过程采用Takara反转录试剂盒实现。使用细菌16S rRNA基因V4区通用引物515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)进行扩增[15],扩增条件为:在94℃条件下高温变性3分钟,后进行30次中温循环,中温循环在94℃条件下持续30 s,56℃条件下持续30 s,在72℃条件下持续40 s,最后在72℃条件下延伸10 min[16]。每个样品获得的PCR产物采用凝胶电泳进行检测,并采用胶提取试剂盒(Sangon Biotech,China)进行提纯,提纯后的PCR按照等摩尔比混合后送测序。
1.3 测序及数据分析
16S rRNA基因扩增子测序和转录组测序在Illumina Hiseq 2000(Illumina Inc,USA)中完成。扩增子测序结果按照QIIME分析流程进行分析[17],原始序列通过标签进行分类,并采用QIIME pipeline对序列质量进行修整,嵌合子序列采用Uchime algorithm进行去除,所得到的高质量序列以97%的序列相似性阈值处理生成OTUs(操作分类单元),并利用RDP(核糖体数据库项目)分类器进行分类分析样品中群落结构。转录组数据在在线数据处理平台—MG-RAST中进行分析。大致流程为:上传到MG-RAST数据库中的FASTQ格式的原始数据,在初步质量控制后,通过“join paired ends”选项将来自于同一样品的两个数据文件合并在一起。测序过程所产生的错误序列通过Gomez-Alvarez等所提到的方法进行去除[18]。低质量的序列通过modified dynamictrim方法进行去除[19]。功能基因的微生物来源分析主要通过将测序数据与MG-RAST中的M5NR数据库进行比对得到。而SEED Subsystems数据库会对蛋白质进一步构建一个代谢通路,被用来进行功能分析。
2 结果与讨论
2.1 氨氮浓度对活性微生物群落结构及丰富度的影响
通过主坐标分析(principal coordinate analysis,PCoA;见图1)研究活性微生物群落结构对不同氨氮浓度的响应。结果显示,R1,R2和R3各个氨氮浓度条件下的微生物聚为一类,表明氨氮浓度对原核微生物群落组成影响较大,不同氨氮浓度反应器种原核微生物群落组成不同。

图1 厌氧发酵系统中基于16S rRNA的主坐标分析
通过对Observed OTUs, Chao 1 指数和PD whole tree指数计算研究原核微生物群落多样性以及某些特定微生物群落活性随氨氮浓度的变化测序深度统一为9190条序列。结果显示微生物多样性指数随氨氮浓度的增加而增大(见表1),且在不同氨氮浓度条件下表现出显著差异。结果表明高氨氮使厌氧系统形成了新的生态位[20]。在高氨氮浓度下微生物种类可能增加,但不同微生物的相对丰度降低,某些微生物失去优势。
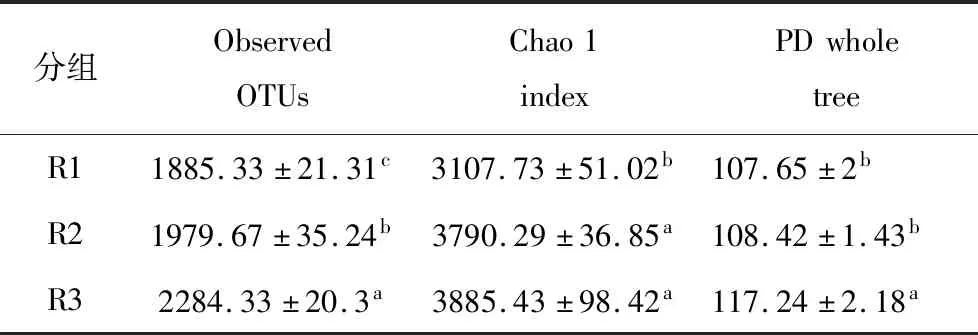
表1 不同氨氮浓度反应器微生物的多样性
注:每行数字后面的字母表示在p=0.05水平上的显著差异
2.2 活性原核微生物群落组成及其表达分析
在本次实验中,通过扩增子测序测定了16S rRNA基因的V4区域,以揭示发酵系统中微生物在门、属水平上的群落组成。结果见表2。

表2 基于16S rRNA主要微生物群落的相对丰度以及与氨氮浓度的相关性
结果显示,厚壁菌门(Firmicutes)的相对丰度随氨氮浓度增加而增加,在R1,R2和R3中相对丰度分别为33.8%,37.5%和53.7%(见表2)。厚壁菌门中的消化链球菌属(Peptostreptococcus),Peptoniphilus,梭菌属(Clostridium),Sporanaerobacter,Tepidimicrobium的相对丰度随氨氮浓度增加而增大,与氨氮浓度呈现显著正相关;而乳酸杆菌属(Lactobacillus),纤维杆菌属(Fibrobacter)和瘤胃球菌属(Ruminococcus)的相对丰度随氨氮浓度增加而降低,与氨氮浓度呈现显著负相关(见表2)。变形菌门(Proteobacteria)的相对丰度随氨氮浓度增加而增大,其代表性属Desulfobulbus与氨氮浓度表现出显著正相关。拟杆菌门(Bacteroidetes)在R2中其相对丰度为最大,为48.2%,而在R1和R3中,其相对丰度相对较低,分别为33.4%和31.1%,其代表性属Ruminofilibacter与氨氮表现出显著负相关。互营菌门(Synergistetes)和螺旋体门(Spirochaetes)及其代表性属与氨氮浓度无显著相关性。互营菌门(Synergistetes)及其主要属Aminobacterium与在R2中的相对丰度达到最大,而螺旋体门(Spirochaetes)及其主要属Treponema表现出相反的变化趋势。

对3组样品总RNA进行转录组测序,研究氨氮对厌氧反应器微生物基因表达的影响。结果见表3。
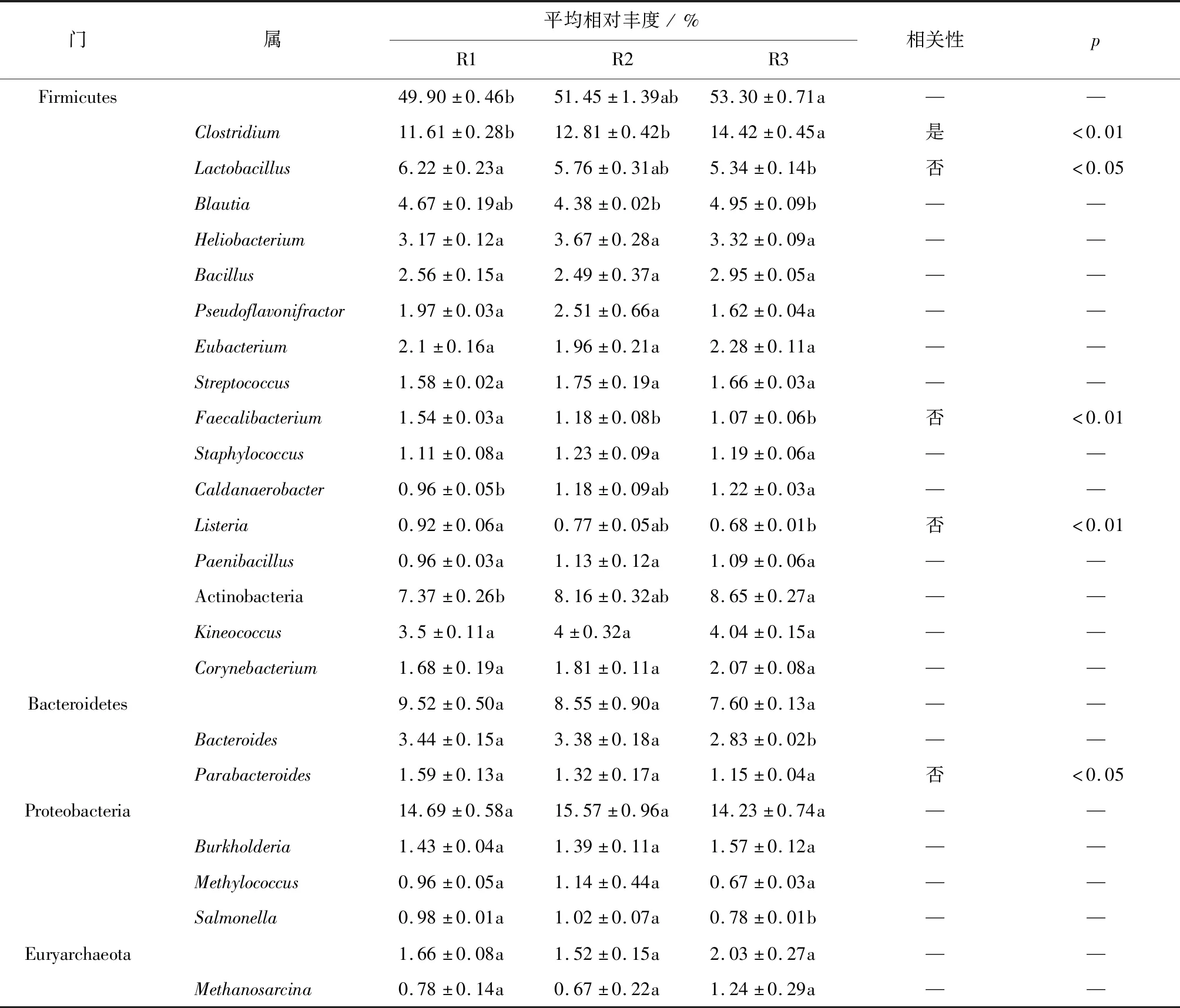
表3 基于转录组数据的主要细菌群落相对丰度及其与氨氮浓度的相关性
对不同功能基因的微生物来源分析结果显示,厚壁菌门(Firmicutes)的功能基因在每个反应器中占据了主要地位(见表3),且其变化趋势与16S rRNA测定结果中的变化趋势相一致,表明高氨氮浓度有利于基因表达的进行,从而使其具有较高的活性。拟杆菌门(Bacteroidetes)的相对丰度随氨氮浓度增加而降低,表明其对氨氮浓度较敏感,基因表达受到抑制。变形菌门(Proteobacteria)在R2中的相对丰度最大,可以看出其对氨氮有一定的耐受性,但氨氮超过一定浓度范围时基因表达受到抑制。门水平上微生物在16S rRNA和mRNA中的差异表明不同微生物在不同氨氮浓度条件下基因的表达情况和代谢活跃程度具有显著的差异。
而转录组数据测定的各个属中,Clostridium,Lactobacillus,Faecalibacterium,Listeria和Parabacteroides随氨氮浓度变化表现出了显著性差异(见表3),其中,只有Clostridium与氨氮浓度成显著正相关,其余属与氨氮浓度成显著负相关。表明氨氮对大多数属的微生物活性基因的表达和代谢活跃程度具有抑制效应,而高氨氮浓度条件下乙酸的积累促进了Clostridium这种转化乙酸、乳酸的微生物的基因表达和代谢活性[23-24]。
2.3 细菌群落的功能基因分析
在MG-RAST数据库的子系统M5NR中对功能基因进行注释[25]。根据这些基因编码蛋白的生物化学功能,我们将所有的功能基因在3个水平(level 1, levle 2, level 3)上进行分组。
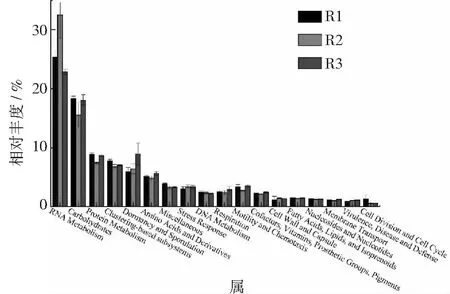
图2 level 1水平上功能基因表达差异。
在level1上(见图2),RNA metabolism的相对丰度最高,在R1,R2和R3中,其相对丰度分别为25.4%,32.5%和22.9%。Carbohydrates,Protein metabolism与Amino Acids and Derivatives具有相同的规律,在R2中相对丰度最小,在R1和R3中的相对丰度较大;Dormancy and Sporulation与Cell Wall and Capsule的相对丰度随着氨氮浓度的增加而增大;而Miscellaneous,DNA Metabolism以及Cell Division and Cell Cycle的相对丰度则随氨氮浓度增加而降低。
RNA Metabolism是分解代谢生化反应基因与酶之间的桥梁,其相对丰度变化趋势表明一定氨氮浓度有利于代谢过程的进行,过高或过低的氨氮浓度会影响反应过程的进行;Carbohydrates,Protein metabolism以及Amino Acids and Derivatives通常存在富含蛋白质和多糖的反应器中[26],其相对丰度变化趋势表明在R1和R3中,氮含量过低或过高,抑制了反应过程的顺利进行,造成了蛋白质和多糖的积累,因此其相应的代谢功能基因增加[27-28];Dormancy and Sporulation与Cell Wall and Capsule属于厚壁菌门,具有与厚壁菌门相同的变化趋势,且该功能基因主要与孢子生殖及细胞壁结构相关[29],结果表明,生殖方式与细胞壁结构会影响微生物对氨氮浓度的耐受性;Miscellaneous、DNA Metabolism以及Cell Division and Cell Cycle的变化趋势则表明高氨氮浓度可能会抑制细胞的形成。

图3 level 2水平上功能水平表达分析
在level2上(见图3),Spore DNA protein,Oxidation stress,Lysine,threonine,methionine and cysteine以及Fermentation的相对丰度均随着氨氮浓度的增加而增大,表明高氨氮浓度影响了细胞的正常生理功能,微生物中的应激反应机制开始启动,各种抑制细胞凋亡的功能蛋白开始增加,以保护微生物免受氧化损伤[30-31];而Plant-Prokaryote DOE project的相对丰度则随着氨氮浓度的增加而降低。Protein biosynthesis,Central carbohydrate metabolism,Monosaccharides,Flagellar motility in prokaryota在R2中的相对丰度较低,表明在氨氮为5500 mg·L-1时有利于反应的进行,该结果与之前的研究结果相一致[32]。

图4 level 3水平上功能水平表达分析
在level3上(见图4),Group Ⅱ intron-associated genes在R1,R2和R3中的相对丰度最高,分别为22.6%,30.2%和20.3%。其次是Small acid-soluble spore proteins,该种功能基因对休眠期芽孢抵抗外界刺激和避免芽孢死亡具有重要作用[33],其相对丰度随氨氮浓度增加而增大;Methanogenesis,Ribosome LSU bacterial的相对丰度则随着氨氮浓度的增加而降低。从这3个水平功能基因的变化趋势可以看出,高氨氮会对发酵过程产生抑制作用,在高氨氮水平下,与微生物应激反应相关的功能基因以及抵抗外界刺激的功能基因的相对丰度增加。
2.4 甲烷生成过程的功能基因分析
通常,产甲烷过程主要有乙酸营养型代谢途径、氢营养型代谢途径和甲基营养型代谢途径[34-35]。转录组数据显示,反应器中甲烷八叠球菌属(Methanosarcina)占主导地位(见表3),且相对丰度随氨氮浓度增加而增大;氢型的甲烷囊菌属(Methanoculleus)次之,其次为甲基和氢混合型的第七产甲烷古菌属(Methanomassillicoccus)[36]。在我们的系统中,氢营养型和乙酸营养型代谢途径的酶基因来自于不同微生物(具有最大相对丰度的编码酶的基因)(见表4)。在乙酸代谢途径中,编码ACK,PTA以及ACS的基因都来源于产甲烷古菌Methanoculleus和Methanosarcina;而编码CDH的基因在对照组中主要来源于Bacteroides,在高氨氮浓度的反应系统中,主要来源于Syntrophomonas。在氢营养代谢途径中,编码各种酶的基因主要来源于产甲烷古菌Methanoculleus和Methanosarcina。
尽管很多研究者认为,在高氨氮条件下,甲烷形成主要通过氢营养代谢途径转化H2和CO2而形成甲烷[37]。然而,在本研究中,乙酸代谢途径与氢营养代谢途径在各个反应器中均存在。乙酸代谢途径中,限制代谢速度的酶ACS在高氨氮条件下相对丰度更大,表明乙酸代谢途径在高氨氮浓度具有更高的代谢活性,与乙酸型的产甲烷古菌相对丰度变化趋势相一致;而氢营养代谢途径中的MCH酶活性在对照组中明显高于高氨氮浓度反应系统中的活性(见表4),表明高氨氮浓度会对氢营养途径形成甲烷具有抑制作用。共同代谢途径的两种酶tetrahydromethanopterin S-methyltransferase (MTR) 和 methyl coenzyme M reductase (MCR) 的相对丰度表现出随氨氮浓度的增加而降低的趋势。在R1和R2反应器中,Methanoculleus是产甲烷古菌中MTR和MCR酶的主要贡献者,但是在R3反应系统中,MTR和MCR酶的主要贡献者则主要是Methanoculleus和Methanosarcina。这些结果表明,高氨氮浓度对产甲烷过程具有抑制作用;不同氨氮浓度条件下,两种产甲烷代谢途径均存在,且乙酸途径占据主导地位。

表4 转录组分析催化乙酸代谢途径和氢营养代谢途径中编码特定酶的相对丰度 (%)
3 结 论
综上,厌氧发酵系统中的高氨氮浓度会改变活性微生物的群落结构和基因表达活性。高氨氮浓度条件下,Ruminofilibacter和Lactobacillus向Clostidium和Peptostreptococcus转变,活性微生物的多样性都显著增加,表明在高氨氮浓度条件下形成了新的特定生态位。高氨氮浓度也会改变功能基因的表达水平,高氨氮浓度条件下与孢子生殖和细胞结构相关的代谢过程基因,如Dormancy and Sporulation和Cell Wall and Capsule会增加,而与细胞生长代谢相关的基因,比如Cell Division and Cell Cycle和Miscellaneous会减少。产甲烷过程中相应酶的转录表达量逐渐减少,表明高氨氮将抑制产甲烷过程的进行,但高氨氮系统中乙酸代谢途径具有更高的代谢活性,乙酸营养型产甲烷途径占据主导地位。这些结果说明,微生物的群落结构和代谢过程会随着氨氮浓度的变化而出现适应性的改变。
猜你喜欢
化工管理(2022年14期)2022-12-02
生物技术进展(2022年5期)2022-10-11
中老年保健(2022年3期)2022-08-24
煤炭转化(2022年4期)2022-07-14
中学生数理化·高一版(2022年4期)2022-05-09
中国沼气(2021年4期)2021-12-15
中学化学(2021年11期)2021-12-09
煤炭转化(2021年4期)2021-07-14
世界有色金属(2021年1期)2021-04-19
中国金属通报(2020年8期)2020-12-31
