
Molecular dynamics study on growth of carbon dioxide and methane hydrate from a seed crystal
2019-12-05 06:28PrajaktaNakateBappaGhoshSubhadipDasSudipRoyRajnishKumar
Prajakta Nakate,Bappa Ghosh,Subhadip Das*,Sudip Roy,*,Rajnish Kumar*
1 Chemical Engineering and Process Development,National Chemical Laboratory,Pune 411008,India
2 Physical and Materials Chemistry Division,National Chemical Laboratory,Pune 411008,India
3 Department of Chemistry,Chaudhary Ranbir Singh University,Jind,Haryana-126102
4 Department of Chemical Engineering,Indian Institute of Technology,Madras,Chennai 600036,Tamil Nadu,India
Keywords:CH4recovery Natural gas hydrate CO2sequestration Kinetics F4order parameter Cage dynamics Thermodynamics
ABSTRACT In the current work,molecular dynamics simulation is employed to understand the intrinsic growth of carbon dioxide and methane hydrate starting from a seed crystal of methane and carbon dioxide respectively.This comparison was carried out because it has relevance to the recovery of methane gas from natural gas hydrate reservoirs by simultaneously sequestering a greenhouse gas like CO2.The seed crystal of carbon dioxide and methane hydrate was allowed to grow from a super-saturated mixture of carbon dioxide or methane molecules in water respectively.Two different concentrations(1:6 and 1:8.5)of CO2/CH4molecules per water molecule were chosen based on gas-water composition in hydrate phase.The molecular level growth as a function of time was investigated by all atomistic molecular dynamics simulation under suitable temperature and pressure range which was well above the hydrate stability zone to ensure significantly faster growth kinetics.The concentration of CO2molecules in water played a significant role in growth kinetics,and it was observed that maximizing the CO2concentration in the aqueous phase may not result in faster growth of CO2hydrate.On the contrary,methane hydrate growth was independent of methane molecule concentration in the aqueous phase.We have validated our results by performing experimental work on carbon dioxide hydrate where it was seen that under conditions appropriate for liquid CO2,the growth for carbon dioxide hydrate was very slow in the beginning.
1.Introduction
Gas hydrates are ice-like crystalline,non-stoichiometric inclusion compounds composed of organic/inorganic moieties which are trapped within the cages formed by a network of hydrogen-bonded water molecules.These water cages are formed by four,five and six membered hydrogen bonded rings which are interconnected via each other to form various types of crystalline structure [1,2].There are three naturally abundant crystalline forms of gas hydrates namely structure I(sI,cubic type),structure II(sII,body-centered cubic type)and hexagonal structure(sH,face-centered cubic type).The sI unit cell has two 512and six 51262cages.Similarly,the sII unit cell has sixteen 512and eight 51264cages,and sH has three 512,two 435663,and one 51268cages[3-6].In the present work,we have dealt with structure I hydrates formed by carbon dioxide and methane independently.
Methane recovery from natural gas hydrate reserves by the injection of industrially emitted carbon dioxide is considered to be a promising approach as it provides the added impetus of simultaneously sequestering extraneous CO2.The method of recovering methane from natural gas hydrates by injection of carbon dioxide is a novel approach [7-10].In 2011,a field trial done by Conoco Philips on Alaska North slope showed that through injection of carbon dioxide,it is possible to produce methane from natural gas hydrates and simultaneously sequester carbon dioxide.Several problems were observed from the field test such as difficulties related to injecting CO2into a less consolidated reservoir at low temperature and production of elevated levels of water and sand.Injection problem was later solved by adding a suitable concentration of N2since it reduced the density in the column[11].However,one of the major concerns for the production of methane from natural gas hydrates by pumping carbon dioxide rich liquid phase was incomplete recovery of methane[12,13].It is evident that optimum temperature and pressure condition necessary to maximize methane recovery is not available.However,for suggesting such thermodynamic conditions at macro scale one needs to understand the kinetics and mechanism of pure methane and pure carbon dioxide hydrate formation at a molecular level.
During hydrate formation,different phenomena e.g.,hydrate nucleation [14,15],cage dynamics in case of mixed hydrate [16],growth mechanism [17,18],kinetics of inhibition and promotion[19-21]occur at a shorter (atomistic scale)time and length scale.Molecular dynamics simulation has been extensively used to probe such events at that time scale and has proven to be an effective tool for detailed quantitative predictions of mechanisms.Walsh et al.[22]has investigated the methane hydrate nucleation and growth by molecular dynamics at suitable thermodynamic conditions of 250 K and 50 MPa.They found the coexistence of sI and sII structures despite there being a thermodynamic preference for sI.This was attributed to the kinetics of formation which governed the nucleation of hydrates involving methane and water.Sarupria and Debenedetti[23]illustrated the homogeneous nucleation of methane hydrate from bulk methanewater in the absence of interfaces.Hydrate like clusters were formed which did not possess the long range order as in methane hydrate.Apart from that,the ratio of the cages in clusters was less than that present in classic hydrate structures.Nada [24]simulated a three phase system of a gas,clathrate,and liquid water in order to investigate the growth mechanism of the clathrate.They reported that growth from the concentrated solution contained some empty cages while no such empty cages were seen in the case of growth from dilute solution.Tung et al.[25]studied the effect of pressure on the growth rate of methane hydrate.They reported that the growth rate depends on methane solubility in the water phase,diffusion of methane by mass transport and the affinity of methane to incomplete water cages at the interface.A significant amount of work has been done on the growth of methane hydrates while the formation and growth study for carbon dioxide hydrates are comparatively rare.Granasy et al.[26]studied nucleation and growth of carbon dioxide hydrates using phase field theory.Tung et al.[27]investigated growth of carbon dioxide hydrates over a wide range of pressures.They reported a new meta-stable medium sized 4151062cage at the hydrate growing interface and subsequent transformation of these cages into more stable sI hydrate.A significant work was done by Zhongjin et al.[28]where they performed microsecond molecular dynamics simulation to deal with carbon dioxide nucleation.They found out that the adsorption of CO2molecules around its own hydration shell is significant for stabilizing the hydrogen bond and hence in inducing the nucleation of CO2hydrate.Sarupria and Debenedetti[29]did another study where they investigated the effects of cage occupancy on the decomposition of CO2hydrate in the presence of amorphous water.They showed that it is important to consider the aspect of relative occupancy in large and small cages rather than the overall hydrate occupancy for studying the effect on CO2hydrate dissociation.
Carbon dioxide can replace the methane from the solid hydrate phase as it is thermodynamically favorable to form CO2hydrate compared to methane hydrate in a subsea environment.However,kinetics of hydrate formation and effect of pressure and resulting liquefaction of CO2on hydrate formation kinetics are not well known.Therefore we believe that,it is important to separately understand the gas hydrate formation from pure methane and pure carbon dioxide respectively.Bulk techniques such as gas uptake measurements are typically used for determining the kinetics of hydrate formation[30].Such measurements suggest a relatively homogeneous process of gradual water to hydrate conversion.However,micro-imaging measurements on hydrate formation of pure methane and carbon dioxide have conclusively shown that at molecular level,conversion of liquid water drops to hydrate is quite inhomogeneous;within a very small reactor,some drops converted to hydrate in few minutes while others required hours or days.Thus quantitative measurements of kinetic processes in sub-volumes of a larger sample suggest that the smaller the volume observed,the more inhomogeneous the process appears to be [31].The intriguing question is how this process of liquid to solid conversion happens in the presence of gases which has very limited solubility in each other.Hydrate may nucleate at the gas-water interface,however,subsequent hydrate growth must involve transport of either water or gas at the interface and it may depend on multiple parameters including the difference in hydrophobicities and by extension,polarities of CO2and CH4.Thus their solubility in water would be very different which would result in different growth kinetics.The consequences of these differences in hydrophobicity and polarity of the guest molecules(CO2and CH4)on hydrate formation kinetics have been addressed in the present work.
We study the growth kinetics of pure carbon dioxide hydrate,and pure methane hydrate starting from a seed crystal under similar thermodynamic conditions.We have maintained a constant temperature and pressure in our simulation box to study the intrinsic growth of hydrate formation.Two different concentrations representing a supersaturated solution of carbon dioxide and methane were studied at multiple temperature and pressure,which was above the three-phase boundary of hydrate formation for achieving sustained hydrate growth.The growth mechanisms of these hydrates were analyzed and growth rates at two different concentrations,temperatures,and pressures were compared.The growth of crystal was monitored using order parameter(discussed in the results section).
2.Simulation procedure
Fig.1 represents a typical hydrate two-phase simulation box,having CO2and CH4seed crystal(solid phase)located approximately in the middle of the simulation cell and CO2or CH4water mixture (liquid phase)on both sides of the hydrate phase in the Z-direction.The hydrate phase used in the study consisted of 3 × 3 × 6 unit cell of structure I hydrate with all its cages filled with CO2or CH4(432 guest molecules and 2484 water molecules).The crystal structure was then randomly solvated with CO2gas molecules (and CH4gas for CH4hydrate)and water on both the sides in the Z-direction to get the final simulation box as shown in ESI(Fig.S1).
MD simulations were carried out by changing the concentration of hydrate forming gas in the aqueous phase.Two different concentrations(1:6 and 1:8.5 mol ratio of CH4/CO2water)used in this study represent a typical gas to water mole ratio for methane and carbon dioxide hydrate respectively.The ratio of 1:6 suggests 1 mole of hydrate forming gas for every 6 moles of water;this concentration is very close to the hydration number of structure I hydrate.Gas molecules were randomly placed in the aqueous phase and the system was equilibrated for a short time at hydrate forming thermodynamic condition (Table 1 for details).The temperature and pressure conditions chosen for this work were above the solid-liquid coexistence line in the three phase diagram of pure methane and CO2hydrate (see ESI fig.S2).After equilibration,it is noted that the simulation box appears quite different for both the systems(CH4and CO2system)in terms of distribution of gases in the aqueous phase,however,to compare the growth kinetics of pure CH4and pure CO2hydrate all the production runs were carried out under same concentration.
Production run simulations were performed using GROMACS 4.5.5 software package[32].The TIP4P/ICE(for water)[33,34],EPM2(for carbon dioxide)[35,36]and OPLS-UA (for methane)[37]forcefield models were considered for all the simulations.The geometric combination rule was used to calculate LJ parameters between the atoms of carbon dioxide-water and methane-water.The detailed force field parameters are given in Table S2.The leapfrog algorithm was used to integrate the equations of motion with a time step of 1 fs for both NVT and NPT runs.The LINCS algorithm was used to constrain the bond length between the atoms in rigid molecules[38].The cut-off distance of 1.2 nm was taken to calculate the short-range non-bonded interactions.Particle Mesh Ewald (PME)summations were used for calculating the coulombic long range interactions with a relative Ewald tolerance of 1×10-6[39,40].

Fig.1.(a)Snapshots of carbon dioxide-water system at the beginning(t=0 ns)and at the end(300 ns)of NPT production run for 1:6 and 1:8.5 ratio of CO2and water.(Cyan—hydrate water,blue—amorphous water,red and yellow—carbon and oxygen of carbon dioxide in hydrate phase,purple and green—carbon and oxygen of carbon dioxide in amorphous phase.)(b)Snapshots of methane-water system at the beginning(t=0 ns)and at the end(300 ns)of NPT production run for 1:6 and 1:8.5 ratio of CH4and water.It is noted that the pressure and temperature conditions were same in both the systems(50 MPa and 260 K).(Red—united atom methane in hydrate phase,green—united atom methane in amorphous phase,cyan—hydrate water,blue—amorphous water.)
The simulation box with periodic boundary conditions was simulated following the procedure described below.Initially,a short equilibration run was carried out for 1 ns in NVT ensemble followed by 5 ns in NPT ensemble with the position restrained hydrate seed crystal to minimize the interfacial energy of solid-liquid boundary.The equilibrated structure was then subjected to the final production run in the NPT ensemble.The Nose-Hoover temperature thermostat [41,42]with a relaxation time of 0.5 ps and Parrinello-Rahman pressure barostat [43,44]with a relaxation time of 1 ps were used for each thermodynamic condition as given in Table 1.Semi-isotropic pressure coupling was used such that the box length scaled along the z-dimension (direction of crystal growth)independent of the x-y direction with a coupling time of 1.00 ps.All the simulations were repeated from scratch(i.e.,building the amorphous water-CO2and water-CH4phase)three times,and the average results were plotted for further analysis.
3.Algorithm for determination of cages
Number of cages during hydrate growth can be monitored through accounting the large and small cages by analyzing the trajectory as proposed by Jacobson et al.[45].The algorithm first looks for all the oxygen atoms which are within 0.61 nm of a guest molecule i.e.,carbon dioxide or methane molecule.In the above criteria it is assumed that a guest molecule which is present anywhere in the cage(at the edge of the cage)will also get counted and be part of the system.With the information on the structure of the water molecules in hydrate form and the topology of the rings they form in structure I hydrate[46],a criterion is selected for the connectivity of oxygen atoms,which is 0.35 nm apart from each other.In the second stage of the algorithm,the search is initiated for possible pentagonal and hexagonal rings which are formed by connecting identified oxygen atoms.The given procedure is used to identify 512and 51262cages composed of 20 and 24 water molecules[47].
4.Results and Discussion
Table 1 summarizes the simulation conditions such as concentration,pressure and temperature used in this study.The pressure andtemperature conditions studied are above the three-phase boundary in the phase diagram of carbon dioxide and water.Figs.S3-S8 in ESI cover the snapshots,of cage formation as a function of time and order parameter plots at different pressure and temperature employed in this study.At varying concentration of guest molecules and thermodynamic conditions,we have selected a subset of relevant conditions and presented in the paper.To start with,we selected a pressure of 50 MPa and temperature of 260 K for further investigation within the stability zone for both CH4and CO2hydrate.Fig.1 shows the snapshots of the production run of CO2and CH4-water systems(Fig.1a and b respectively)performed at 260 K and 50 MPa with the different concentrations of guest gases (1:6 and 1:8.5).As can be seen in Fig.1a,the aqueous phase is well distributed at the beginning of the NPT production run.While at the end of the production run (after 300 ns),the growth of well-arranged hydrate layers on the seed crystal can be observed.Fig.1b shows the production run of CH4-water system.As seen in the figure,CH4molecules form clusters in the aqueous phase at the start of the production run(0 ns)which is mainly attributed to the hydrophobicity of methane molecules.As shown in the snapshot with both the concentrations(1:6 and 1:8.5),approximately six layers of hydrate have formed on the top of the seed crystal.The partial densities of all the production runs have been presented in the ESI(Fig.S9)which is in good concurrence with the snapshots.Fig.2 shows the number of cages as a function of time for carbon dioxide and methane hydrate respectively at 260 K and 50 MPa.It is quite clear that all the four systems show gradual hydrate growth albeit at different rates.On comparing Fig.2(a)and(b),it is clear that there is a sharp rise in the cage count when we decrease the concentration of carbon dioxide molecules to 1:8.5 which can also be clearly adjudged from the snapshots in Fig.1(a)and (b).Whereas,the rate of hydrate growth (or the cage count)at different concentrations of methane shows no significant difference as can be seen from Fig.2(b).The orderness(or extent of hydrate growth)can also be quantified by the use of four body structural order parameter(F4).It denotes whether the phase is in the hydrate region or in the aqueous phase.

Table 1 Systems studied in this work
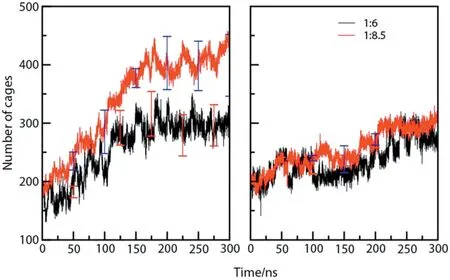
Fig.2.Evolution of cages(total 512 and 51262)during simulation.Panel(a)denotes growth of carbon dioxide hydrate and (b)denotes growth of methane hydrate.The data was obtained at a temperature and pressure of 260 K and 50 MPa.
F4is defined ascos3Ø where,n is the number of oxygenoxygen pairs of water molecules present within 0.3 nm of distance from each other and ø is the torsion angle between them.The average value of F4for hydrate system and water is 0.7 and-0.04 respectively [48].Fig.3 represents the F4order parameter trend with respect to time at 260 K and 50 MPa for both 1:6 and 1:8.5 concentrations of carbon dioxide and methane.The simulation box was divided in equal layers based on the length of a unit cell in order to study the layerwise change in F4order parameter.The layer denominations are given in ESI as Fig.S10.A typical increase in the order parameter in Fig.3(a),has a confirmation of the region proceeding towards the hydrate phase.The region which is just besides the hydrate seed crystal i.e.,Layer 1 (red)on both the sides of hydrate seed crystal shows an increase in the order parameter before it progresses to the second layer adjacent to the first layer and so on.This confirms the hypothesis that hydrate growth proceeds in a layerwise fashion[49,50].From Fig.3(a),it can be seen that at lower CO2concentration,an additional layer of hydrate is formed within the same time frame of 300 ns.Similar trend was also observed for methane hydrate growth (Fig.3(b))however the overall hydrate growth of CO2hydrate is faster than that of methane hydrate.
As discussed above,all the systems studied above had a tendency for growth towards the hydrate phase at favorable thermodynamic conditions.Interestingly,for some system,no hydrate growth was observed.Fig.4(a)shows a snapshot of such a system at 270 K and 50 MPa with a concentration ratio of 1:6 of carbon dioxide molecules at 300 ns.Fig.4(b)shows a snapshot of methane hydrate at the same thermodynamic conditions for comparison and it shows sustained hydrate growth with time.At 270 K,even at a pressure as high as 50 MPa,the hydrate phase shows no hydrate growth for CO2system.But,if one focuses on methane hydrate at the same thermodynamic conditions in Fig.4(b),barring a few layers,the hydrate phase growth is complete.This can also be easily inferred if we look at Fig.5 which denotes the evolution of total number of cages with respect to time.Considering the error bar,the number of cages more or less remains constant with time for the growth of carbon dioxide hydrate while it increases in the case of methane hydrate growth.This unique feature of stunted hydrate growth of CO2hydrate at such a high pressure and low temperature can be explained suitably.The factor pertains to the mass transfer limitation in the case of four phase system(liquid water,hydrate water,liquid CO2and gaseous CO2).Under the conditions given(270 K,50 MPa and 1:6 initial concentration of CO2),occurrence of CO2-CO2clustering is favorable compared to growth of CO2hydrate.Due to the presence of liquid CO2and given that the simulation timescale is significantly small,no hydrate growth was observed.To prove the hypothesis,we have conducted experiments wherein the incoming supply of carbon dioxide for hydrate growth is in two phases,one with gaseous CO2and another with liquid CO2.The results of the experiments are plotted in terms of pressure variation in Fig.6.A pressure drop is seen due to the enclathration of gas molecules in the solid hydrate phase.Rate of the drop in pressure is directly proportional to rate of hydrate growth.As can be observed from Fig.6,the rate of hydrate formation with liquid CO2is gradual compared to that with gaseous CO2.That is,the hydrate formation is slow and is completed in about 10 h from the nucleation point(for liquid CO2).Whereas the hydrate formation for gaseous CO2is completed in about 2.8 h from the onset of nucleation.

Fig.3.Order parameter(a)of carbon dioxide hydrate and(b)of methane hydrate plotted in a layerwise fashion at a temperature and pressure of 260 K and 50 MPa.Left and right side denotes the left and right side of the seed crystal in the Z-direction of the simulation box.
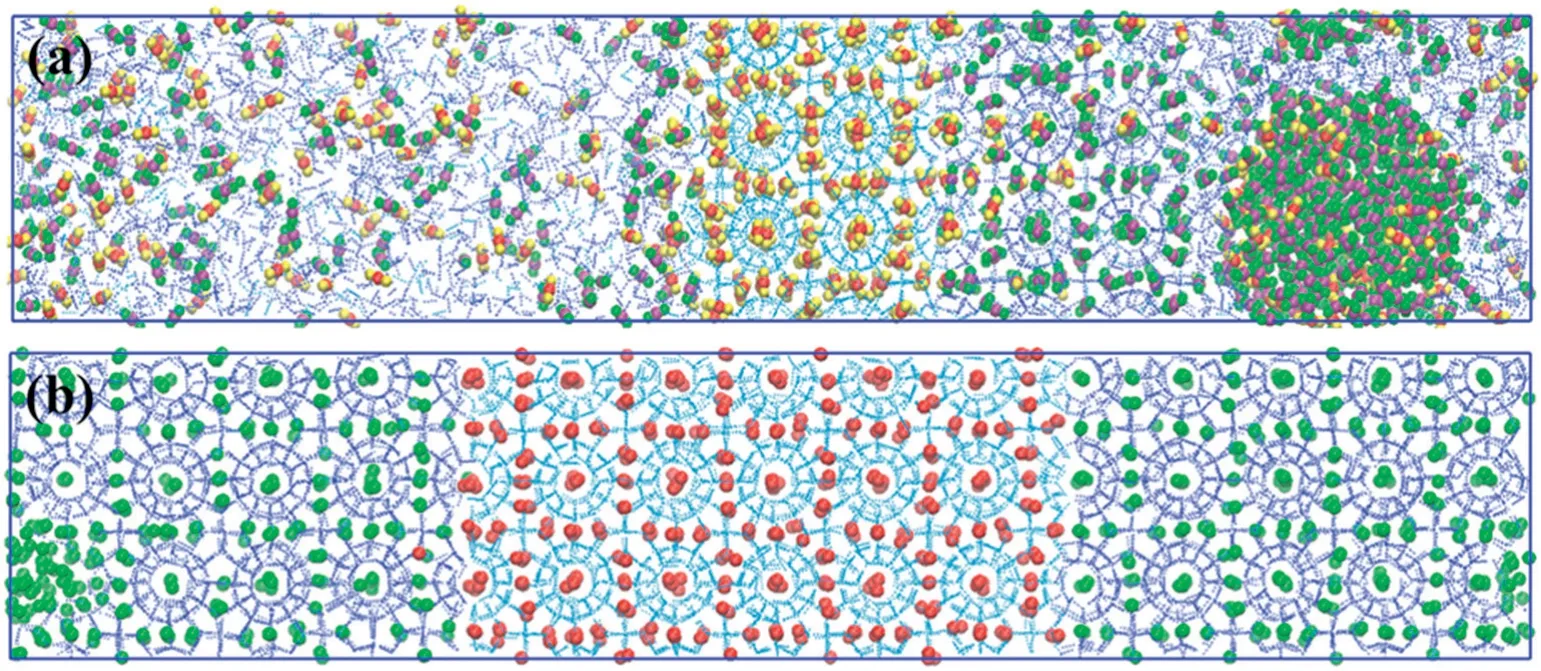
Fig.4.(a)Snapshot of a system at 300 ns with 1:6 concentration of carbon dioxide molecules to water at 270 K and 50 MPa.(Cyan—hydrate water,blue—amorphous water,red and yellow—carbon and oxygen of carbon dioxide in hydrate phase,purple and green—carbon and oxygen of carbon dioxide in amorphous phase.)(b)Snapshot of a system at 300 ns with 1:6 concentration of methane molecules to water at 270 K and 50 MPa.(Red—united atom methane in hydrate phase,green—united atom methane in amorphous phase,cyan—hydrate water,blue—amorphous water.)
Idea behind this work was to identify if the intrinsic kinetics of methane hydrate formation and carbon dioxide formation are of similar time scale or there are certain differences.One may perform experiments in the laboratory setup and compare this,however,the three phase hydrate equilibria of these two systems differ significantly.Thus,it is very difficult to decouple the gas uptake data related to kinetics(arising through the heat and mass transfer limitation)and the thermodynamics(which basically affects the hydrate growth by a factor(Pexp-Pequ)).Pexprefers to the experimental pressure at which hydrate growth kinetics is studied,Pequrefers to the three phase boundary,and this is the minimum pressure at which a gas-liquid system would convert into a gas-liquid-solid system.The difference Pexp-Pequis also known as driving force.Higher pressure in the simulations was chosen for two reasons,at sufficiently high pressure the driving force(Pexp-Pequ)is going to be almost same for both the system,CO2/CH4.Also,the kinetics of hydrate growth would be faster at higher pressure and thus would save computational time.As discussed,in the experiment,the aim was to keep CO2in liquid phase and simulate the possibility of CO2-CO2interaction as speculated in the simulation.This was possible around the pressure given(3.6 MPa)and hence we were able to qualitatively compare across the experimental and simulation conditions.
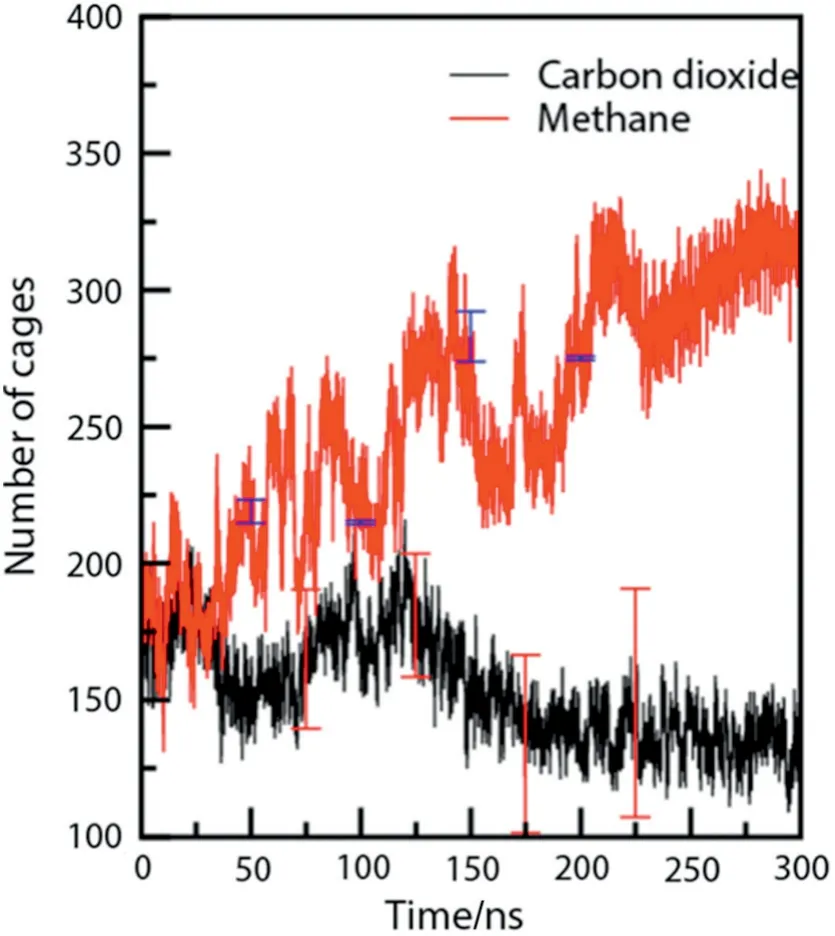
Fig.5.Evolution of cages(total 512 and 51262)during simulation.The black and red curve is at a temperature and pressure of 270 K 50 MPa and a concentration of 1:6 with respect to water.
5.Conclusions
In this work,we have studied the intrinsic growth of carbon dioxide and methane hydrate from a seed crystal as a starting point.Growth is studied at different thermodynamic conditions with the combination of two temperatures (260 K &270 K)and a pressure of 50 MPa.Two suitable concentrations of hydrateforming gas in the aqueous phase (1:6,1:8.5)are then chosen for NPT ensemble as a production run for molecular dynamics simulation.A total of sixteen systems were investigated for the growth of carbon dioxide and methane hydrate.Based on the four body structural order parameter(F4)and the number of cages which originated during the growth process we witnessed sustained growth of the seed crystal for all the methane hydrate system (irrespective of methane concentration in the aqueous phase).
However,with the system involving the growth of carbon dioxide hydrate,hydrate growth rate decreased with respect to the increase of concentration of the guest molecules in the aqueous phase.Moreover,at 50 MPa and 270 K the system with a higher concentration ratio of 1:6(gas to water)witnessed no hydrate growth.It was observed that at these thermodynamic conditions,CO2clustering was favored over hydrate growth.At lower CO2to water concentration (1:8.5),the trend of hydrate growth was on expected line and hydrate continued to grow with time as expected.An investigation was done to reveal the reason behind this behavior.We carried out experiments pertaining to incoming flow of gaseous and liquid CO2and observe its effects on the growth of CO2hydrate.We saw that the rate of hydrate growth is clearly dependent on the state of CO2that is supplied.The above result clearly shows that replacing methane from natural gas hydrate reservoir requires an optimum initial concentration of CO2in the aqueous phase so that the phase separation is not predominant for the desired result.
Acknowledgments

Fig.6.Hydrate growth(a)and dissociation curves(b)for CO2hydrate under conditions resembling gaseous and liquid CO2.
We sincerely thank Dr.Prithvi Raj Pandey a former PhD student in the group for his valuable guidance and discussion.SD would like to thank CSIR,India,for providing his PhD fellowship.We greatly acknowledge the computational support given by CSIR Fourth Paradigm Institute(CSIR-4PI)Bangalore for providing computational resources and support.We acknowledge the support of other sponsored project in the group from which the computational facility was built which came handy during this work.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2019.02.006.
 Chinese Journal of Chemical Engineering2019年9期
Chinese Journal of Chemical Engineering2019年9期
- Chinese Journal of Chemical Engineering的其它文章
- Decomposition behaviors of methane hydrate in porous media below the ice melting point by depressurization☆
- Research progress in hydrate-based technologies and processes in China:A review☆
- Fundamental mechanisms and phenomena of clathrate hydrate nucleation☆
- Methane hydrates:A future clean energy resource
- Progress and trends in hydrate based desalination(HBD)technology:A review☆
- Extraction of methane hydrate energy by carbon dioxide injection-key challenges and a paradigm shift