
Rh-ZSM-5分子筛吸附CO、H2O和O2的理论研究
2019-11-29 10:25:18吴安安
厦门大学学报(自然科学版) 2019年6期
陈 丹,吴安安,谭 凯,吕 鑫
(厦门大学化学化工学院,福建省理论与计算化学重点实验室,福建 厦门 361005)
过渡金属铑(Rh)基催化剂已广泛应用于多相催化的各项研究[1-3].最近,文献报道在有CO与O2共同存在时,沸石负载的单核铑催化剂(Rh-ZSM-5)可在423 K温和的液相反应中将甲烷直接转化为乙酸和甲醇,且CO在反应中具有不可或缺的作用[4-5].其中,Tang等[4]根据详细的能谱研究数据认为初始催化剂Rh-ZSM-5具有[Rh1O5]的配位环境;Shan等[5]以CO为探针分子,运用漫反射红外傅里叶变换光谱(CO-DRIFTS)测得该反应条件下吸附CO的特征振动峰值为2 116和2 049 cm-1,并归属于单核双羰基铑Rh(CO)2物种的对称与反对称伸缩振动.
大量研究表明CO可吸附在单原子级分散表面Rh+上形成羰基铑化合物.Yang等[6]最早发现CO吸附在Rh/Al2O3上在红外光谱中会出现两个特征峰,归属于双羰基铑Rh(CO)2表面物种的对称和反对称伸缩振动.Ivanova等[7]进一步发现,室温下ZSM-5负载的Rh位点可与不同配比的CO结合形成单至三羰基铑Rh(CO)n(n=1,2,3)表面物种,红外光谱特征振动峰值分别为2 114 cm-1(Rh(CO)),2 114和2 048 cm-1(Rh(CO)2),2 181、2 118和2 084 cm-1(Rh(CO)3).大量的光谱数据测得Rh(CO)2的红外对称与反对称伸缩振动频率范围分别为2 120~2 075 cm-1和2 053~1 990 cm-1[8-10].理论研究表明,CO与其他小分子(H2、N2、C2H4和NO等)可共同吸附在Rh-ZSM-5簇上形成铑配位化合物,且CO与Rh位点间具有较强的配位作用[11-13].据此推测,在最近报道的Rh-ZSM-5催化甲烷转化反应中[4-5],CO会与其他小分子形成共吸附物种而非简单的单核双羰基铑Rh(CO)2物种,且CO等吸附物种的强配位还可能会影响到表面Rh原子与ZSM-5基底的配位成键,因此,有必要就此开展更为系统而细致的理论研究,以期为后续Rh-ZSM-5催化甲烷部分氧化反应机理的理论研究建立合理的模型基础.
本文中采用含18个硅氧四面体(以下简称18T)的簇模型来模拟Rh-ZSM-5分子筛上Rh+的配位环境,运用密度泛函理论(DFT)方法计算了Rh-ZSM-5上小分子CO、H2O和O2吸附和共吸附物种的结构和能量;为更好地关联相关实验研究[4-5]结果,分别研究在423 K下有、无CO共存时各吸附物种的稳定性.结果表明:当H2O或CO的吸附分子数达到饱和时,Rh+中心与ZSM-5基底的配位数由3降至1;H2O和O2共存体系中形成的最稳定表面吸附物种为Rh(H2O)2(O2);而一旦引入强配位能力的CO,最稳定的表面吸附物种变为Rh(CO)2(H2O),阻碍了O2的化学吸附,暗示具有活化甲烷C—H键能力的活性氧物种并非如先前人们所预期的由O2的化学吸附[5]所致.
1 计算方法与模型
本研究全部的理论计算均使用杂化密度泛函B3LYP方法[14-16]在Gaussian 09程序包[17]中完成.由于文献中一般采用包含相对论效应赝势的基组SDD或LanL2DZ对Rh原子进行计算[18-20],所以本研究选取赝势基组SDD对Rh原子进行计算[21-22],其余原子(Si、Al、O、C及H)则均采用6-31G(d,p)基组[23-24];对各吸附态构型进行几何优化和振动频率分析,以确保优化构型为稳态结构,同时获得零点能(ZPE)校正值和吉布斯自由能(G423 K),能量单位均为kJ/mol.因同一结构可能有不同自旋态,本文中所讨论的每个结构的左上方均标注其自旋多重度.吸附自由能的计算公式为Gad=Gcomplex-(GRh-ZSM-5+Gadsorbate).
由于分子筛体系较大,所以簇模型是用量子化学方法来研究分子筛的常用方法之一.本研究从ZSM-5分子筛的晶格[25]中选取一个18T的分子筛簇模型(图1(a)),模型截断时边界Si原子上的悬挂键由H原子饱和且位置固定(Si—H键长约为0.148 nm,方向保持与分子筛中原有的Si—O键方向一致),其余原子的坐标则未加任何限制(图1(b)).该簇模型中的T12位为已知的最优Al3+取代位[26-28],为保持电中性,其紧邻的Al—O—Si桥氧通常吸附一个质子(即所谓的B酸位,见图1(a)~(b)),Rh+与质子交换,即构成Rh-ZSM-5分子筛催化剂[4-5]的表面簇模型,以此为初始结构进行优化,得到3种不同的Rh+结合位点(图1(c)).

(a) ZSM-5;(b) ZSM-5的18T簇模型;(c) Rh+在ZSM-5内可能吸附位点的构型.图1 ZSM-5簇模型Fig.1 Cluster model of ZSM-5
2 结果与讨论
2.1 Rh-ZSM-5中Rh与表面原子的成键
首先考察了Rh+与ZSM-5孔道表面原子的结合方式及其可能电子态,包括开壳层三重态、闭壳层单重态和开壳层单重态,发现在ZSM-5孔道内Rh+结合的3个最可能位点(图1(c))的基态均为三重态(表1),Rh+与表面原子的配位数分别为3,3,2,且至少与Al3+紧邻的2个桥氧配位;注意到基态下3种结合方式的自由能差小于21.0 kJ/mol,因此在较高温度下Rh+可以在3种吸附位点动态迁移.且因Rh+在位点1的锚定在能量上最优,故对应的3Rh-ZSM-5构型将作为研究小分子CO、H2O和O2吸附的参考零点.

表1 Rh+吸附结构各低能电子态的ΔG423 K及Rh—O键长
2.2 H2O吸附
计算表明3Rh-ZSM-5表面簇的Rh+位点上最多可同时吸附3个H2O分子,吸附1~3个H2O分子的过程保持自旋守恒,吸附体系的基态均为三重态(图2(a)~(c)),吸附过程放热,总吸附自由能分别为-43.1,-54.0和-77.8 kJ/mol.由于3Rh-ZSM-5的Rh+位点本身配位不饱和,所以吸附第1个H2O分子时放热最为剧烈,随着吸附H2O分子数量的增多,Rh+保持了四配位,即H2O分子吸附实质上只是取代表面氧原子的配位,Rh+与表面原子的配位数依序降低,最终变为1(图2(c)).
2.3 O2吸附及其与H2O共吸附
O2分子基态为三重态,吸附到3Rh-ZSM-5的Rh+上后,吸附体系基态为开壳层单重态,吸附模式为侧式,吸附较稳定(图2(d)),吸附分子O—O键长为0.130 nm,为超氧(O2-)形态,同时表面金属离子由Rh+变为Rh2+;吸附自由能为-55.2 kJ/mol,比单个H2O分子的吸附在能量上更有利.进一步计算发现,第2个O2分子在Rh+上只能发生物理吸附,在此不作赘述.需要指出的是,图2(d)所给出的O2化学吸附态正是Tang等[4]计算研究甲烷部分氧化机理的前驱态,但注意到此时Rh仍然与3个表面桥氧配位,因此,在甲烷部分氧化的水相环境中,也极可能发生与H2O分子共吸附取代部分表面桥氧配位.
计算表明,共吸附1个H2O分子(图2(e))时Rh+与表面桥氧的配位数已降低到1,但O2仍保持与Rh+的侧式配位,总吸附自由能降低到-85.8 kJ/mol,即1个H2O分子的共吸附为放热过程.共吸附2个H2O分子(图2(f))时放热更为明显,总吸附自由能为-97.5 kJ/mol;同时吸附的O2分子尽管仍呈超氧态(O—O键长为0.130 nm),但已转变为端位吸附模式,自旋未成对电子主要定域于外端氧原子,在几何和电子因素两个方面均有利于外端氧原子与其他反应物分子的反应.因此,在有H2O共存时,考察O2分子的吸附活化及其后续参与甲烷部分氧化的过程时,不应忽视H2O分子共吸附对反应机理和反应能学的影响,此前的研究中以类似图2(d)的简单表面活性氧物种为前驱物研究甲烷部分氧化机理[4]的做法值得商榷.
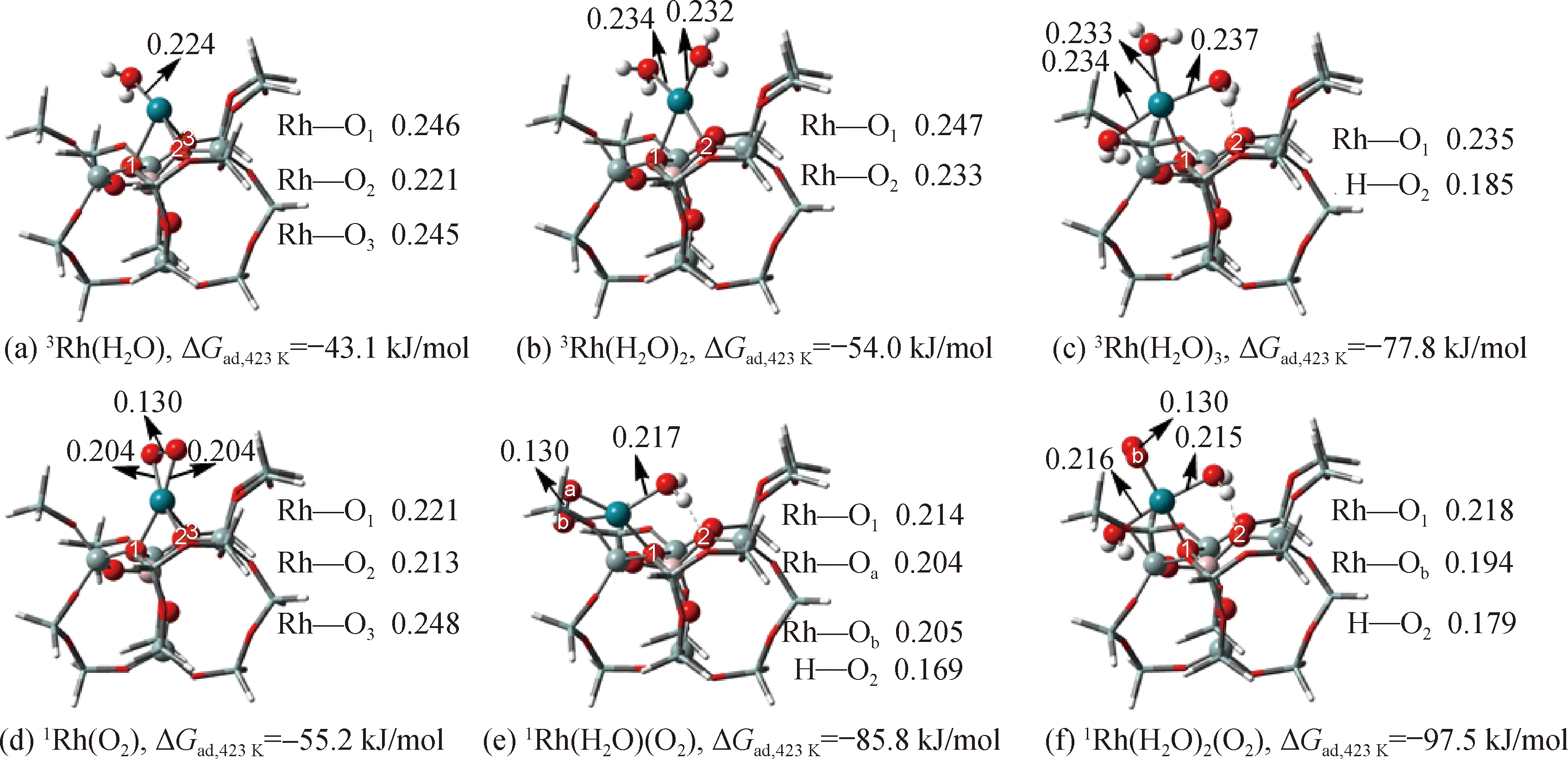
图2 H2O与O2在Rh-ZSM-5上的吸附构型和吸附自由能(键长单位:nm)Fig.2 Configurations and adsorption free energies of H2O and O2 adsorbed on Rh-ZSM-5 (bond length in nm)
2.4 CO吸附及其与H2O、O2共吸附
不同于H2O和O2,CO是配位能力强的优良配体,在金属有机化学中发挥了重要作用,在过渡金属中心上的配位、吸附或解离时常引起金属中心电子态的变化[29-30].因此,对CO在3Rh-ZSM-5表面簇上吸附所形成的吸附体系,本研究分别考察了三重态和单重态两类电子态(图3).
1个CO分子吸附在3Rh-ZSM-5表面簇的Rh+上时,吸附体系的基态仍为三重态(图3(a)),对应的吸附自由能为-126.8 kJ/mol,比单重态(图3(b))的自由能低50.2 kJ/mol,说明CO在Rh+上的吸附能力远强于H2O和O2,因此在三者共存时,必须优先考虑CO吸附,而这一点在此前对Rh-ZSM-5催化甲烷部分氧化机理的理论研究[5]中恰恰被忽视了.

图3 CO在Rh-ZSM-5上的吸附构型和吸附自由能(键长单位:nm)Fig.3 Configurations and adsorption free energies of CO adsorbed on Rh-ZSM-5 (bond length in nm)
2个CO分子吸附在Rh+上后,吸附体系的三重态(图3(c))反而比单重态(图3(d))的自由能高110.9 kJ/mol,即吸附体系基态变为单重态,总吸附自由能为-242.7 kJ/mol,说明CO配体与金属的配位作用不仅很强,而且还改变了Rh+金属中心电子自旋态.这种Rh(CO)2表面物种在之前对CO/Rh-ZSM-5吸附体系的理论和实验研究[6-10]中均有报道.
3个CO分子吸附体系的三重态(图3(e))也比其单重态(图3(f))基态的自由能高87.0 kJ/mol,其单重态基态的总吸附自由能(-243.5 kJ/mol)与2个CO分子吸附体系的几乎相当,说明在CO/Rh-ZSM-5吸附体系中,Rh(CO)3和Rh(CO)2表面物种共存.这2个表面物种中Rh+均保持四配位,与H2O/Rh-ZSM-5吸附体系中的金属中心配位数一样.
注意到CO/Rh-ZSM-5吸附体系中第3个CO分子的吸附为热中性,因此,以H2O取代第3个CO分子来作为(CO+H2O)共吸附模型,得到Rh(CO)2(H2O)表面物种的2个异构体(图4(a)和(b))的总吸附自由能(-271.1和-248.9 kJ/mol)均比Rh(CO)3体系低,说明有H2O共存时,(CO+H2O)的共吸附反而会优于三羰基吸附物种;这种更为有利的(CO+H2O)共吸附应该与共吸附表面物种Rh(CO)2(H2O)中同时存在吸附H2O与ZSM-5晶格氧之间较强的氢键作用(HOH…O—Al氢键键长为0.167 nm左右,图4(a)和(b))有关.相较而言,在Rh(CO)3表面物种中再引入的1个H2O分子未能与Rh+形成配键,而是通过氢键吸附在表面桥氧上(图4(c)和(d)),在能量学上均颇为不利,对应的H2O分子吸附自由能分别为2.9和26.8 kJ/mol.因此,在Rh-ZSM-5催化甲烷部分氧化反应[4-5]中因CO和H2O共存,它们在Rh+中心上的配位共吸附形成如Rh(CO)2(H2O)的稳定表面物种,在反应机理研究中不应被忽视.

图4 CO、H2O和O2在Rh-ZSM-5上的共吸附构型和吸附自由能(键长单位:nm)Fig.4 Configurations and adsorption free energies of CO, H2O and O2 co-adsorbed on Rh-ZSM-5 (bond length in nm)
本研究还进一步考察了O2与CO和H2O在Rh-ZSM-5上的共吸附(图4(e)和(f)),发现在Rh(CO)2(H2O)表面物种上,O2只能物理吸附(图4(e));而三者直接配位共吸附于Rh+上(图4(f))在能量学上也颇为不利,总吸附自由能比Rh(CO)2(H2O)混合吸附体系高108.8 kJ/mol.这说明在三者的混合体系中不太可能通过配体取代方式形成O2活化前驱物.
上述理论结果说明,Rh-ZSM-5催化甲烷部分氧化反应[4-5]中,因CO和H2O共存发生强混合吸附,O2在Rh+上的活化以及形成具有活化甲烷C—H键能力的活性氧物种,绝非简单的O2分子化学吸附[4]所致,必须审慎考虑(CO+H2O)共吸附物种在其中扮演的角色及其与O2反应生成活性氧物种的可能性.
2.5 1 Rh(CO)2(H2O)化合物的红外光谱
进一步计算了羰基表面物种1Rh(CO)2以及共吸附表面物种1Rh(CO)2(H2O)的红外振动频率,其中振动频率的标度因子为0.970 2[31],计算得到自由CO的C—O伸缩振动频率为2 143 cm-1,1Rh(CO)2物种中C—O的对称与反对称伸缩振动频率为2 105和2 046 cm-1,1Rh(CO)2(H2O)物种中C—O的对称与反对称伸缩振动频率为2 109和2 051 cm-1,与Shan等[5]对CO/Rh-ZSM-5吸附体系在常温大气环境中的实验观测峰值(2 116和2 049 cm-1)吻合.
3 结 论
本文中对小分子CO、H2O和O2在Rh-ZSM-5上的吸附及共吸附进行了详细的理论探讨,结果表明:1) 小分子H2O和CO均易吸附于表面Rh+中心,形成稳定的四配位表面物种,同时也会影响Rh+中心与ZSM-5基底的配位,吸附分子饱和配位后Rh+与基底的配位数降至1;2) 相对于CO和O2,H2O在Rh+中心的吸附能力较弱,却能与O2或CO形成稳定的共吸附物种;3) 仅有H2O和O2共存时,最稳定的表面吸附物种为Rh(H2O)2(O2);4)引入强配位能力的CO后,最稳定的表面物种为变为Rh(CO)2(H2O),阻碍了O2的化学吸附.本研究为进一步对Rh-ZSM-5催化甲烷部分氧化反应机理开展更为全面而细致的理论研究建立了合理的模型基础.
猜你喜欢
数学物理学报(2022年3期)2022-05-25 13:33:22
数学物理学报(2022年1期)2022-03-16 06:15:04
数学物理学报(2021年5期)2021-11-19 07:01:16
数学物理学报(2021年3期)2021-07-19 06:02:18
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
当代陕西(2019年6期)2019-04-17 05:04:10
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
无机化学学报(2014年4期)2014-02-28 17:31:08
