
腺苷酸活化蛋白激酶在纤维化发生中的作用*
2019-11-21 05:14:18李淑娴于功昌
中国病理生理杂志 2019年11期
李淑娴, 贾 强, 于功昌△, 邵 华△
[1济南大学山东省医学科学院医学与生命科学学院, 2山东省职业卫生与职业病防治研究院, 山东第一医科大学(山东省医学科学院), 山东 济南 250062]
[中图分类号]R363; R364.5[文献标志码]A
doi:10.3969/j.issn.1000- 4718.2019.11.028
1 AMPK的结构和生物学作用
腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)被认为是调节能量稳态和代谢应激反应的能量传感器,是一种进化保守的丝氨酸/苏氨酸激酶,由调节性β(β1和β2)和γ(γ1、γ2和γ3)亚单位以及催化性α(α1和α2)亚单位组成[1],其中α亚基含有催化核心的N端激酶结构域(kinase domain,KD),Thr172 位点的磷酸化可使AMPK活性增加超过100倍,被认为是AMPK活化的标志。C末端结合β、γ亚基并含有重要的调节结构域,即当AMP水平下降时,可以抑制AMPK活化的自身抑制结构域(autoinhibitory domain,AID)和富含丝氨酸/苏氨酸的结构域“ST环”(ST-loop)[2-3];β亚基的N端有碳水化合物结合模块(carbohydrate-binding module,CBM)将AMPK固定在细胞膜上,而N端区域之后紧跟着α、γ结合区域[4],因此,β 亚基相当于1个与 α 和 γ 亚基结合的支架,使AMPK形成稳定的异源三聚体[5];γ亚基的N末端包含4个串联的胱硫醚β-合成酶(cystathionine β-synthase,CBS)重复序列,形成2个可以结合AMP或ATP分子的Bateman结构域[6]。 AMPK 3个亚基的分子结构如图1所示。目前认为AMPK的激活方式主要有以下3种:(1)AMP/ATP相关的变构调节机制:AMP通过连接到 γ 亚基变构激活AMPK[7];(2)AMPK活性的自调节:AMPK复合物的 α 亚基C端含有自抑制序列,能够抑制AMPK的激活;(3)AMPK激酶激活:主要有钙离子/钙调素依赖性蛋白激酶β(calcium/calmodulin-dependent protein kinase β, CaMKK-β)、转化生长因子β激活蛋白激酶1(transforming growth factor beta-activated protein kinase 1,TAK1)和肝激酶 B1(liver kinase B1,LKB1)[8]。当细胞内能量缺失时AMPK被激活,激活后的AMPK通过减少ATP消耗途径和增加ATP的代谢途径来维持能量代谢平衡,从而使体内ATP的总量增加或比例增加[9]。然而,AMPK不仅能调节能量代谢,还有促进自噬、诱导凋亡、调节氧化应激和抗炎症等作用。
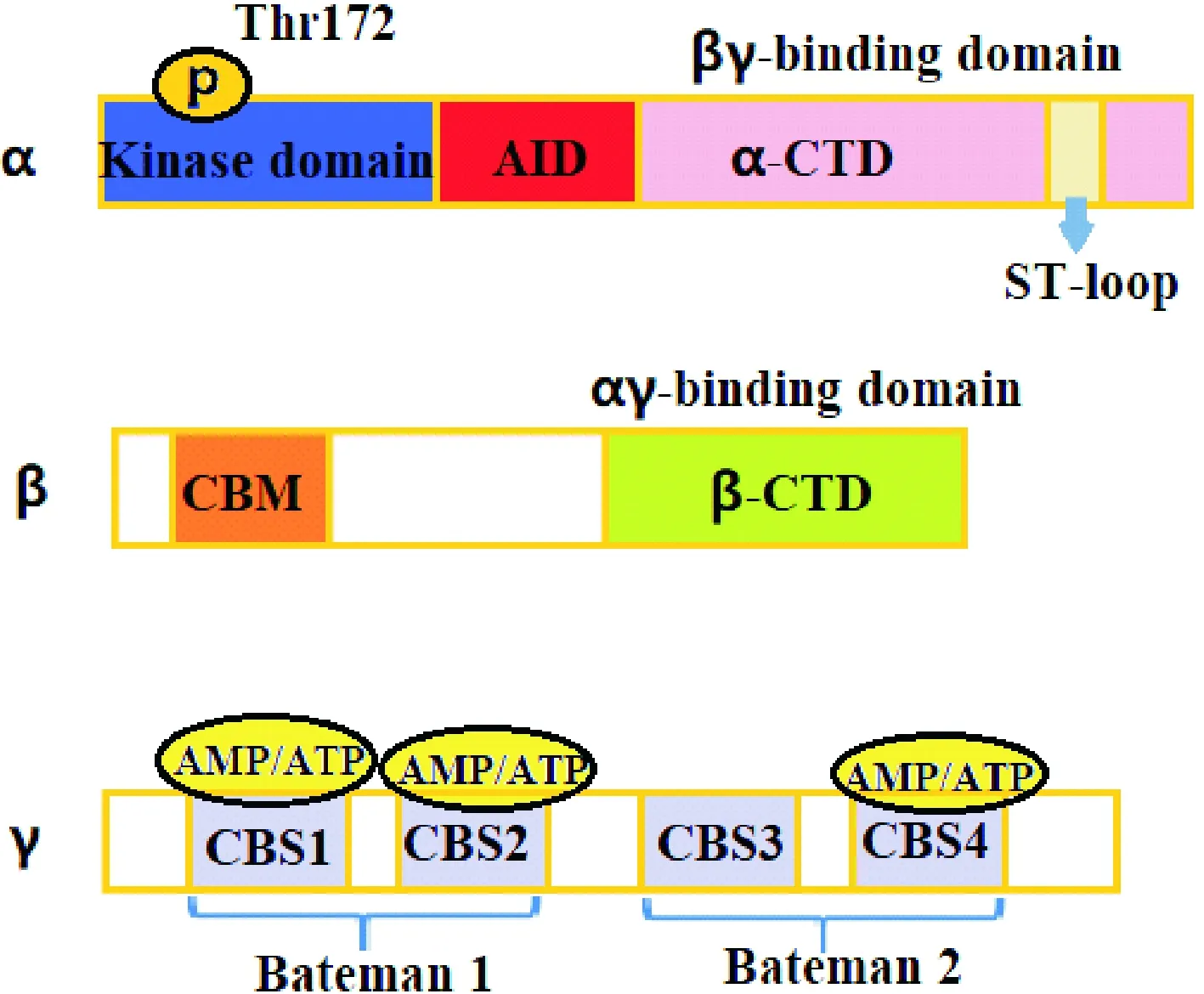
Figure 1.The structure of the 3 subunits of AMPK. AID: autoinhibitory domain; CBM: carbohydrate-binding module; CBS: cystathionine β-synthase.
图1 AMPK的3种亚基结构示意图
2 AMPK与炎症反应
纤维化发生过程是一个复杂的病理生理学过程。首先,损伤刺激触发免疫细胞的快速募集,导致炎症反应,产生大量促炎细胞因子和生长因子,然后炎症细胞因子激活效应细胞并促进其增殖,触发肌成纤维细胞的活化[10-11]。在诱导纤维化的因素中,Th2细胞释放与纤维化发展相关的白细胞介素4 (interleukin-4, IL-4)、IL-5和IL-13。IL-13具有致炎和免疫调节的作用,可以作用于成纤维细胞表面IL-4R/IL-13Rα1而激活成纤维细胞,并在特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)的发生、发展中通过促进巨噬细胞的激活从而在促纤维化过程中发挥重要作用[12-13]。单核细胞和巨噬细胞在组织重塑和纤维化中起着至关重要的作用,巨噬细胞来源的转化生长因子β(transforming growth factor-β, TGF-β)诱导成纤维细胞迁移,促进成纤维细胞生长、活化和胶原合成[14]。大量证据表明,AMPK在炎症调节中发挥重要作用。在肺纤维化、慢性阻塞性肺疾病及肺损伤模型大鼠中,多种药物通过激活AMPK途径减少或降低炎症因子[如肿瘤坏死因子α(tumor necrosis factor-α, TNF-α)和IL-6]并减少炎症细胞数量以减轻肺部炎症反应[15]。TGF-β是肝脏炎症损伤的关键调节因子,AMPK可以抑制TGF-β诱导的纤维化。Lim等[16]用TGF-β处理人(LX-2)和大鼠(CFSC-2G)肝星状细胞(hepatic stellate cells,HSC),发现AMPK减弱TGF-β诱导的与转录辅激活剂p300的Sd3相互作用,并改善纤维化激活。慢性炎症和纤维化是Duchenne肌营养不良症的特征,AMPK的激活可以抑制促炎巨噬细胞ltbp4的表达,从而导致TGF-β1分泌减少,刺激促炎巨噬细胞向抗炎状态的转移,减轻病变部位纤维化程度[17]。AMPK的抗炎作用多伴随着转录因子NF-κB的抑制,活化的AMPK主要通过下游蛋白如沉默交配型信息调节物2同系物1(silent mating type information regulation 2 homolog 1,SIRT1)、叉头盒O3a(forkhead box O3a,FoxO3a)、p53、过氧化物酶体增殖物激活受体γ辅激活因子1α(peroxisome proliferator-activated receptor γ coactivator 1α,PGC-1α)等间接抑制NF-κB活性,并降低炎性因子表达[18]。由此可知,AMPK可通过抑制巨噬细胞来源的TGF-β的分泌,也可通过间接调节 NF-κB活性和抑制炎性因子表达来减轻纤维化。
3 AMPK与上皮-间充质转化(epithelial-mesenchymal transition,EMT)
EMT在胚胎发育、伤口愈合、组织再生、肿瘤进展和器官纤维化等多种生物过程中起关键作用[19]。EMT的特征是细胞间接触丧失、基底膜的破坏、蛋白质(如E-钙黏蛋白)的丧失、细胞骨架的重组,以及获得新的间充质标志物[如α-平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)、波形蛋白、I型胶原蛋白和纤连蛋白]而转变为纺锤形形态[20]。随着对纤维化机制研究的深入,近年来发现肾、肝、肺、眼和其它器官的纤维化都有EMT的发生[21]。TGF-β、表皮生长因子(epidermal growth factor, EGF)、胰岛素样生长因子1(insulin-like growth factor-1,IGF-1)和血管内皮生长因子(vascular endothelial growth factor,VEGF)水平的变化会影响正常和异常情况下的组织稳态,触发EMT和进行性纤维化[22]。众所周知,TGF-β调节结缔组织生长因子(connective tissue growth factor,CTGF)和IL-6,在EMT中起重要作用。AMPK通过抑制Smad2/3磷酸化和转录活性来抑制TGF-β诱导的EMT标志物的表达,因此,AMPK激活可抑制TGF-β调节的EMT[23]。哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)被认为是AMPK的下游靶点。Dong等[24]研究表明,二甲双胍和白藜芦醇可以通过激AMPK/mTOR信号来缓解细胞衰老和EMT。AMPK通过直接磷酸化mTORC1受体(mTOR的调节相关蛋白)来抑制mTORC1的活性,从而调节自噬,进而逆转EMT[25]。综上所述,AMPK主要是通过抑制mTOR活性以及抑制TGF-β而阻止EMT进程,从而抑制纤维化发生发展。
4 AMPK与细胞外基质
纤维化的特征是细胞外基质(extracellular matrix,ECM)的合成增加,降解相对不足,随着纤维化进展,组织成分发生变化,如α-SMA和波形蛋白表达增加,E-钙黏蛋白丢失[19]。不同病因的损伤会导致复杂的级联反应,包括多种细胞类型和分子信号,导致ECM的过度积累,从而导致纤维化[26]。目前普遍认为,器官纤维化的发生机制与TGF-β的过表达导致ECM过度沉积且降解不足密切相关[27]。TGF-β1可通过激活经典Smad途径和非Smad的信号途径诱导肾纤维化,从而导致肌成纤维细胞的活化,ECM的过量产生和降解抑制[28]。心脏纤维化的特征是间质成纤维细胞增殖以及包括胶原和纤连蛋白在内的心肌ECM的过度产生和沉积。最近的一项研究揭示了激活AMPK可以通过调节TGF-β介导的Smad3依赖转录来抑制血管紧张素Ⅱ诱导的心脏纤维化[29]。另外,增强自噬也可以减少ECM沉积。Patel等[30]发现沉默自噬相关基因LC3-B和beclin-1后,α-SMA和纤连蛋白等肌成纤维细胞标志物表达明显增多,说明抑制自噬能促进肌成纤维细胞增殖和ECM沉积。研究表明,AMPK激活可以通过增强自噬,同时下调ECM蛋白的稳态水平,从而重新编程IPF成纤维细胞的代谢[31]。因此,AMPK可通过调节TGF-β以及增强自噬,从而减少ECM沉积,调节纤维化。
5 AMPK与肌成纤维细胞
肌成纤维细胞是肺纤维化的发病机制和进展过程中细胞外基质的主要来源,兼具成纤维细胞和平滑肌细胞的特性。研究发现,在肺纤维化的发病机制中,肌成纤维细胞的凋亡受到抑制,导致纤维化进行性发展[32]。在各种促纤维化细胞因子中,TGF-β是已知的最有效的肌成纤维细胞分化诱导剂[33]。在IPF中诱导的肌成纤维细胞分化表现为肺成纤维细胞中I型胶原和α-SMA表达水平的增加[31]。现已经证明,TGF-β1和Smads被认为是肾纤维化的治疗靶点[34]。TGF-β1激活下游Smads信号传导,使Smad2/3复合物磷酸化并将细胞溶质Smad4转运到细胞核中以调节纤维化相关基因表达,重组细胞骨架成分,诱导向肌成纤维细胞转化[35]。除了典型的TGF-β/Smads途径之外,TGF-β1还通过非Smad信号途径诱导肌成纤维细胞活化和肾纤维化,包括丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)、磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶 B(AKT)和小GTP酶途径等[36]。实验证明,TGF-β诱导的肌成纤维细胞分化能够被二甲双胍通过活化AMPK介导的机制有效抑制[37]。在心脏成纤维细胞中,TGF-β1是损伤后成纤维细胞活化/分化的主要决定因素,AMPK激活可以抑制TGF-β介导的肌成纤维细胞的分化[38]。因此,AMPK激活可通过抑制TGF-β/Smads途径,抑制肌成纤维细胞凋亡,从而减弱纤维化。
6 AMPK与纤维化的治疗
有许多药物包括阿卡地新、二甲双胍、白藜芦醇、他汀类药物和小檗碱等通过对AMPK的激活减轻纤维化。AMPK激活剂如5-氨基咪唑-4-甲酰胺核苷酸(5-aminoimidazole-4-carbox-amide-1-β-D-ribofuranoside,AICAR)在肝纤维化动物模型中显示抗纤维化作用[39]。体外实验表明,AICAR处理后,原代大鼠HSC中TGF-β、α-SMA和I型胶原蛋白的基因表达水平显著降低[40]。AICAR可以通过被腺苷转运体转运进入细胞,在细胞内被腺苷激酶酸化生成AMP类似物——ZMP而激活AMPK[41]。二甲双胍是一种用于治疗Ⅱ型糖尿病的药物,可抑制TGF-β1的产生,并通过抑制线粒体呼吸链复合体I和ATP的合成激活AMPK抑制心脏纤维化[42]。二甲双胍治疗还能抑制纤维化发生相关蛋白的表达,降低Smad2/3和细胞外信号调节激酶1/2的磷酸化[43]。白藜芦醇是一种多酚,具有有效的抗氧化和抗肿瘤活性以及对神经系统的重要保护作用。最近Lan等[44]证明白藜芦醇通过至少2种机制激活AMPK。其一为抑制ATP合成,其二为Sirt1-LKB1激活的能量依赖机制。另外,他汀类药物也被证明可以用来治疗纤维化。Reichert等[45]实验证明他汀类药物减轻了大鼠心肌梗死后的纤维化反应和炎症反应。因此,AMPK激活剂对减轻纤维化发挥着重要的作用,AMPK可能会成为一种治疗纤维化的有希望的药物靶点。
7 总结与展望
综上所述,AMPK可以通过抑制TGF-β分泌、调节NF-κB活性和抑制炎性因子发挥抗炎作用;通过抑制mTOR活性以及抑制TGF-β阻止EMT进程;调节TGF-β以及增强自噬,从而减少ECM的沉积;通过抑制TGF-β/Smads途径抑制肌成纤维细胞分化,减弱纤维化。应用AMPK激活剂可以有效控制纤维化。AMPK有望成为纤维化新的治疗靶点。这为治疗纤维化提供了新的方向。然而,AMPK的具体抗纤维化作用机制尚未完全探索清楚,关于AMPK抗纤维化的下游靶蛋白并未完全明确。动物和细胞实验研究发现AMPK激活剂可以对纤维化产生作用,但是,在临床上是否可以治疗纤维化还需进一步验证。因此,进一步探究AMPK的具体抗纤维化机制,明确下游靶蛋白,验证AMPK激活剂是否能治疗纤维化,对于减轻及治疗纤维化有重要意义。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13 08:59:50
天津医科大学学报(2021年3期)2021-07-21 09:03:46
云南医药(2021年3期)2021-07-21 05:40:30
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
中国当代医药(2015年33期)2015-03-01 02:09:20
中国医学科学院学报(2013年6期)2013-03-11 20:26:04
