
姜黄素分光光度法测定水中硼的优化检测条件研究
2019-11-15 09:33邢书才杨永岳亚萍
中国测试 2019年6期
关键词:分光光度法
邢书才 杨永 岳亚萍

摘要:针对姜黄素光度法测定水中硼分析方法存在的问题,研究和优化分析测定的实验条件。采用方法研究的方式,对样品显色反应的蒸发条件进行改良,用普通烘箱替代水浴条件,对样品进行显色和蒸发处理;同时,通过对分析方法校准曲线标准点的重新设置,拓宽方法测定范围,也使校准曲线的线性以及精密度得以显著提高。补充试剂空白试验,增加测定下限处的标准点;并从分析测定质量控制角度,对校准曲线制备中,0.10~1.00mL标准溶液过小的加入量进行改进,变为统一体积量的一致性加入,减小了分析误差的几率。研究结果表明,改良后的实验条件,分析方法的检出限为0.019mg/L,测定下限为0.076mg/L;测定范围为0.080~1.4mg/L,比改进前拓宽了40%。精密度测定结果相对标准偏差小于3.0%,准确度的检验结果符合方法学的技术要求。改进后的分析方法,测定下限低,范围广,灵敏度高;适合于水和废水中硼的分析测定。
关键词:硼;姜黄素;分光光度法;测定条件
中图分类号:O657.3;X832 文献标志码:A 文章编号:1674-5124(2019)06-0065-05
收稿日期:2019-01-22;收到修改稿日期:2019-03-05
基金项目:生态环境部国家标准制修订项目(2018-16)
作者简介:邢书才(1958-),男,北京市人,教授级高级工程师,主要从事环境监测和国家标准的研究。
0 引言
分光光度法測定水和废水中硼,是环境监测和分析检测实验室普遍采用的分析方法。现行检测硼的标准分析方法,主要有GB/T5750.5-2006《生活饮用水标准检验方法无机非重金属指标》和HJ/T49-1999《水质硼的测定姜黄素分光光度法》[1-2]。其中姜黄素分光光度法作为国家环境保护标准分析方法,已经实施了近20年;在分析测定应用中,显现出该方法操作较为繁琐,实验条件比较苛刻,需在无水条件下进行。同时存在校准曲线的试剂空白缺失,测定下限与实际情况偏离较远,测定范围过于狭窄,以及样品和校准曲线制备中取样量过小,易于产生分析误差等问题;这些因素都不同程度地影响到分析测定的质量。分光光度法测定硼的分析实验,以往已有一些文献报道[3-11],对于姜黄素光度法测定水中硼的研究,文献报道较少,且主要涉及分析方法的应用和方法比对方面[12-15];对分析方法中存在问题的改进性研究,近年来未见有报道。本工作针对姜黄素分光光度法存在的问题,进行了方法改进和针对性研究;简化了样品显色和蒸发装置,补充了试剂空白,降低了测定下限,拓展了分析方法的测定范围;实验操作质量控制方面的改进,改善了校准曲线线性不理想、回归系数较差的问题,提高了校准曲线的精密度;使分析测定的实验条件得到优化,提高了分析测定的精密度和准确度,取得了满意结果。
1 实验部分
1.1 要设备
Cary300UV-Vis型光谱仪(安捷伦);塑料烧杯、试剂瓶、25ml成套具塞(PP)塑料比色管,日本NIKKO公司生产;移液管、容量瓶(PP材质,Brand);瓷蒸发皿;恒温水浴槽;恒温烘箱。所有器皿不得使用含硼玻璃材质器皿。
1.2 主要试剂
硼标准贮备液(1000mg/L)(钢研纳克,AccuStandard);硼标准使用液(1.00mg/L,10.00mg/L);硼标准样品(GSB07-1979-2005):盐酸,草酸,姜黄素,无水乙醇(AR,上海化学试剂有限公司)。
姜黄素+草酸溶液:于80mL无水乙醇中加入草酸5.0g及姜黄素0.040g,溶解后再加入浓盐酸4.2mL,移入100mL容量瓶用乙醇定至刻度。
1.3 实验方法
1.3.1 校准曲线的制备
1)原方法:按照HJ/T49-1999《水质硼的测定姜黄素分光光度法》的实验步骤,配制各标准点浓度溶液,测定后制备校准曲线。测定体系的浓度按照总体积为1.00mL计算。
操作方法:向一系列蒸发皿中分别加入0.20,0.40,0.60,0.80,1.00mL(1.00μg/m1)的硼标准使用液,并分别加入0.80,0.60,0.40,0.20mL去离子水,使溶液总量为1mL,分别加入姜黄素-草酸溶液4.0ml,,将蒸发皿轻轻摇动使溶液混匀,置蒸发皿于(55±3)℃的水浴上蒸发至干,并继续于水浴上保留15min后取下蒸发皿,放置至室温后,用少量乙醇溶解并转入25ml,比色管中,用乙醇稀释至刻度,离心后,用2cm比色皿,于波长540nm处,以纯水为参比,测定吸光度。
2)改进法:参照HJ/T49-1999《水质硼的测定姜黄素分光光度法》的条件步骤,改变标准使用液的加入方法,最终用一致性体积的加入方法,制备校准系列溶液;加入显色剂后改用在(55±1)℃的烘箱中蒸发至干,然后按分析步骤进行测定,绘制校准曲线。测定体系的浓度按照总体积为1.00mL计算。
操作方法:在7个100mL容量瓶中,分别加入10.00μg/mL的硼中间液0.00,1.00,2.00,4.00,8.00,12.00,14.00mL,以纯水定容摇匀,1~7号容量瓶中硼的浓度为0.00,0.10,0.20,0.40,0.80,1.20,1.40μg/mL;各取1.00mL于1~7号蒸发皿中,1~7号蒸发皿中含有硼的质量为0.00,0.10,0.20,0.40,0.80,1.20,1.40μg,分别加入姜黄素-草酸溶液4.0mL,将蒸发皿轻轻旋动使溶液混匀,然后将蒸发皿置于(55±1)℃的烘箱中蒸发至干,继续保持15min,取出蒸发皿,冷却至室温,用少量乙醇溶解后转入25mL比色管中,用乙醇稀释至刻度,离心后,用2cm比色皿,于波长540mm处,以纯水为参比,测定吸光度。
1.3.2 样品的测定
1)原方法:吸取样品1.0mL(硼含量不超过1.0μg)于蒸发皿中,以下按1.3.1的1)的实验程序测定,将吸光值带入1.3.1的1)校准曲线中计算出硼浓度值。
2)改进法:吸取样品1.0mL(硼含量不超过1.4μg),按1.3.1的2)实验程序测试,把吸光值带入1.3.1的2)校准曲线中计算出硼浓度值。
2 结果与讨论
2.1 检定下限与检定范围的改良
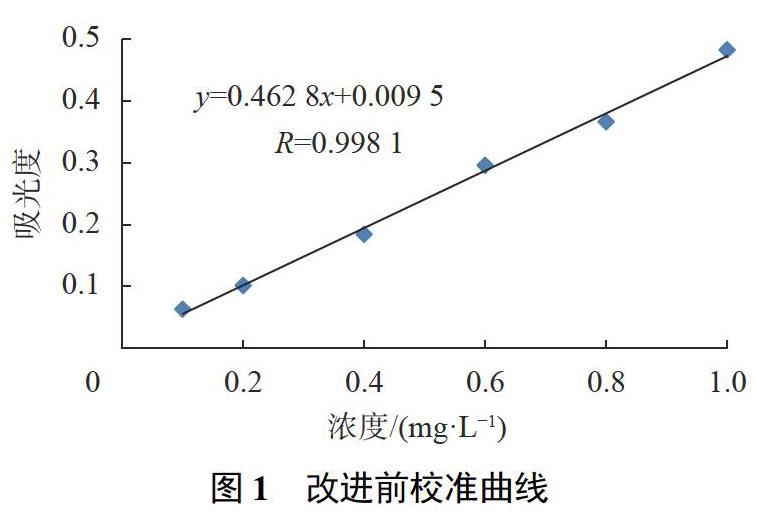
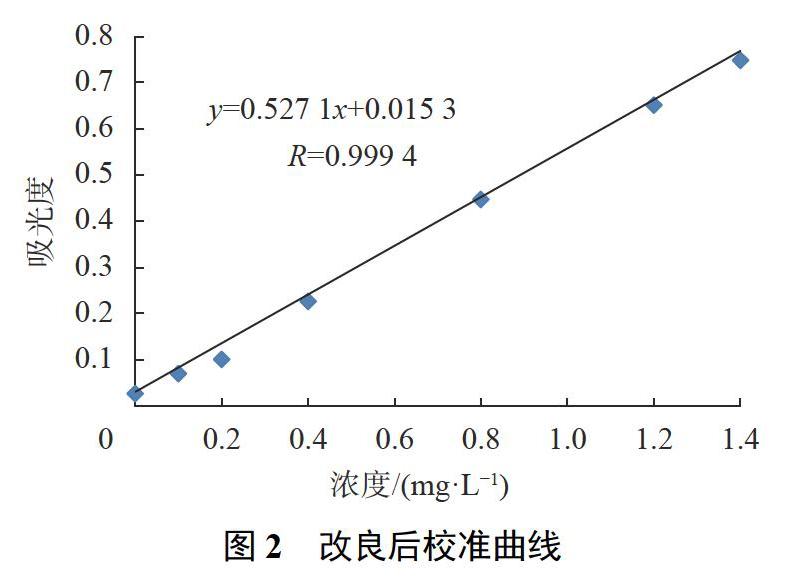
对改进前及改进后的检测方法制备的系列浓度标准点进行测定,并分别制备校准曲线(如图1,图2所示)进行比对分析。
按分析方法标准制修订技术导则的相关规定[16],分析方法的检出限可由校准曲线的的参数计算:检出限=0.01×b-1(检测下限为4倍的检出限)。由此可计算出原方法的检出限和检测下限分别为0.022mg/L和0.086mg/L;改良后方法的检出限和检测下限分别为0.019mg/L和0.076mg/L,较原方法降低了近14%左右。
从检测范围上,改进前后方法的检测范围分别为0.10~1.0mg/L和0.080~1.4mg/L,改良后校准曲线线性良好,精密度较高,检测范围较原方法拓宽了40%之多。
2.2 蒸发装置和质量控制的改进
由图1可见,改进前方法的校准曲线,线性欠佳,数次重复性实验,线性均不理想。原因有多方面,实验过程操作较为繁琐复杂,校准曲线制备过程取样量过小,以及被测样品取样量过小都蕴含分析误差因素。实验中样品所需蒸发时间,蒸发的速度,脱水等条件,都要求有较高的一致性程度。前消解中稍有操作不当容易造成样品损失甚至实验失败的结果。同时,冗长复杂的操作以及显色基团稳定性原因,都可能造成实验分析误差。
以取样量误差分析,原方法校准系列的取样量为0.20,0.40,0.60,0.80,1.00mL,还要分别补水至1.00mL的过程。以1.00mL为例,在取样过程如果出现1滴溶液(约0.05mL)的损失,造成的相对误差为5%。相对于0.20,0.40,0.60mL,将会造成更大的误差。为了控制取样量的误差因素,本研究预先配制与校准系列浓度相同的系列浓度标准使用液,采用在7个蒸发皿中一致性加入1.00mL标准使用液的方法,对取样量的误差因素进行控制(详见1.3.12))。从图2可见,经重复性比对实验,改进后分析方法的校准曲线,从线性质量和精密度上都有了较大提高。
同时,对方法实验的蒸发装置进行了改进。原分析方法是利用水浴控制(55±3)℃,对样品进行显色和蒸发脱水。现改为在烘箱中进行这一步骤,其优点在于相对于水浴,空气的热传导是多方向传导,可使样品反应受热更均匀,温度更加易于控制;样品置于烘箱的托盘之中,相对于水浴,更加有利于样品的安全性,也相对降低了实验成本,更有利于大批量样品的处理。
2.3 校准曲线标准点设置上的改进
对于一种相对测定的分析方法,分光光度法的校准曲线,其中标准点的设置有特定的规则和技术上的要求。根据精密度要求,标准浓度点在布置上要中间范围稀少,而在曲线的两端相对性地密集设置,改良前校准曲线在标准点的分布上,采用的是等均性方式设定(见图1);改良后的校准曲线采用的是中部疏散两端密集的布置方式,并根据实际测定下限的测定值设定定量下限浓度点,有利于低浓度样品的测定;益于校准曲线的质量及精密度的提高,符合分析测定质量控制和方法学要求[16-17]。实际测定结果也表明,相对于原测定方法,无论是校准曲线的线性指标,还是曲线的精密度都有了显著性提高,为分析方法的准确测定提供了技术上的支持和基础。
2.4 试剂空白的补充改进
对于分析方法研究,主要涉及分析方法的精密度、准确度和空白分析。可见空白试验是分析方法研究的一个不可缺少的方面。对于分光光度法,是一种相对测定方法,就其试剂空白而言,不仅关系到测定平台问题,也可以通过试剂空白预知和判断试剂干扰情况,也就是分析判断测定体系背景干扰的大小。如上所述,分光光度法作为一种相对测定方法,参比溶液是其中的测定平台,试剂空白往往受到操作环境、仪器状态以及试剂等多方面因素的影响,其中以试剂造成的影响为主。实际测定中,有的分析方法使用纯水为参比,有些分析方法使用试剂空白为参比溶液。在一个实验分析测定过程中,什么情况下使用纯水为参比,什么情况下使用试剂空白为参比,要依具体情况而定。而对试剂空白的测定情况,就是判断以纯水还是试剂空白为参比溶液的重要依据。
姜黄素分光光度法测定水中硼,使用纯水为参比溶液,在校准曲线的制备过程中,校准系列中的试剂空白是缺失的。所带来的问题是在实验分析中,无法根据试剂空白的大小判断背景干扰情况,也不利于确认参比溶液的选择或参比溶液的灵活选择。本实验在原测定方法中增加了测定的试剂空白实验,使测定方法中的缺失趋于完整,也使测定方法在整体上更加趋于合理。
经实际重复测定,姜黄素光度法测定水中硼的试剂空白值,吸光度在0.026~0.040左右,试剂空白值稍大,有一定的背景干扰。《水和废水监测分析方法(第四版)》相关方法[1B],使用试剂空白为参比;HJ/T49-1999《水质硼的测定姜黄素分光光度法》使用纯水为参比。由于考虑到试剂空白值较高,同时考虑到实验是在乙醇介质中进行,乙醇的易挥发性影响试剂空白作为参比时的稳定性,易使比色皿中参比液的透光率發生改变,使分析测定受到影响;经实际测定,60min后,试剂空白的吸光值变化了0.004,而纯水未发生变化;相对而言,纯水的稳定性是肯定而相对持续的。因此,在这种前提条件下,以纯水为参比是一种较为适宜的实验条件选择。
2.5 精密度和准确度测定
按照方法研究的基本要求[16],用硼标准贮备液配制(高、中、低)3种不同浓度样品后进行实际测定,用以验证改良方法精密度情况。准确度的检查,用水质硼标准样品进行实际测定的验证。测定结果分别汇列在表1、表2中。
从表1列出的测定结果可见,对高、中、低3种浓度样品的测定结果显示,测定结果与样品值一致性较好,相对标准偏差最大值小于3%;说明分析方法的精密度良好。
由表2对硼标准样品的测定数据可以看出,原测定方法和改进后测定方法的测定均值,分别为1.05mg/L和1.03mg/L,分別用t检验法与标准值进行检验,检验结果t原方法=2.31,t改进法=1.40,均小于临界值t(0.05,5)=2.45。说明测定结果与标准值没有显著性差异。同时对改进前和改进后方法测定结果的均值进行t检验,检验值t实验=137
猜你喜欢
农业科技与装备(2016年8期)2017-03-09
中西医结合心血管病电子杂志(2016年25期)2017-03-03
江苏农业科学(2017年1期)2017-02-27
科技视界(2016年9期)2016-04-26
科技资讯(2015年19期)2015-10-09
科技与创新(2015年2期)2015-02-11
