
基于简化基因组技术的云南蓝果树群体遗传分析
2019-11-15 08:05:26张珊珊康洪梅杨文忠
植物研究 2019年6期
张珊珊 康洪梅 杨文忠
(云南省林业科学院,云南省森林植物培育与开发利用重点实验室/国家林业局云南珍稀濒特森林植物保护和繁育国家林业局重点实验室,昆明 650201)
云南蓝果树是典型的极小种群野生植物[1]。目前,对云南蓝果树开展了一系列的濒危机理研究,在系统分类、形态修订、种子萌发特性、抗逆生理及生殖生物学等方面做了大量研究工作[2~6],探索并实践了云南蓝果树就地保护、近地保护、迁地保护、种苗繁育和回归引种等系列的保护措施[7~8]。然而,这些实践究竟保护了多少遗传资源,基于遗传管理的保护措施是否科学有效是目前亟需解决的问题。要想解决这个问题,首先要弄清楚云南蓝果树野生植株的遗传多样性和遗传结构如何。
群体遗传学是研究群体的遗传结构及其变化规律的遗传学分支学科[9]。在群体遗传学的研究中,遗传多样性的高低是一个重要的指标,反映了物种应对栖息地变化的适应性和改变能力,而遗传多样性在群体间的分布,即遗传结构是受许多因素作用的,另外还与物种自身的生物学特征和进化史有着密切关系[10~11]。因此,我们在保护遗传资源时需要先了解居群的遗传结构和遗传背景,进而再制定合理的保护政策[12~14]。
目前最先进的第二代DNA测序技术可通过产生大量的分子标记数据进行成株的群体遗传多样性分析,建立详细的遗传档案[15]。基于酶切的简化基因组测序(RAD-Seq,Restriction-site Associated DNA Sequence)是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序,可大幅降低基因组的复杂度,降低建库和测序成本,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,实现遗传进化分析及重要性状候选基因的预测[15~17]。RAD-Seq可以检测基因组上未知变异点中新的SNP,发掘新的和稀有的变异,对解决群体遗传学[18]、遗传图谱构建[19]、功能基因挖掘[20]、群体进化[21]等问题,具有重大的科研和产业价值。
因此,本研究采用基于二代测序技术的RAD-seq方法对云南蓝果树开展成株的群体遗传分析,为每个成株建立详细的遗传档案,用SNP位点刻画其遗传特征,完成系统进化树、群体结构、PCA分析,从基因组水平揭示不同个体之间的遗传分化关系,进而为遗传资源的保护提供理论基础。
1 材料与方法
1.1 试验材料
对云南蓝果树分布区域现有的24个天然植株进行野外取样,在详细记录植株编号的同时,采集新鲜嫩叶利用液氮快速冷冻并拍照,带回实验室放入-85℃冰箱备用。
1.2 DNA提取与质量控制
采用Sigma试剂盒提取云南蓝果树的基因组DNA。用1%琼脂糖电泳和分光光度计Nano Drop 2000C(Thermo Scientific,USA)检测所提样品的DNA质量,以保证提取的DNA光吸收值A260/280介于1.7~2.1、样品浓度>30 μg·L-1、单个样品总量>1 μg。
1.3 文库构建
文库构建方法流程如下:(1)稀释基因组DNA样品;(2)根据选定的酶切方案(EcoRⅠ),对检测合格的各样品基因组DNA分别配制酶切反应体系,在Thermomixer上适温反应一定时间酶切基因组DNA;(3)配制P1 index adapter连接反应体系,在Thermomixer中适温反应一定时间使P1 index adapter与酶切产物连接;(4)将P1连接产物按照适当的比例进行pooling;(5)配制打断体系,使用打断仪对pooling产物进行打断,再使用试剂盒对打断产物进行纯化;(6)用琼脂凝胶电泳对产物进行片段选择;(7)配制末端修复反应体系,在Thermomixer中适温反应一定时间对片段选择后的产物进行末端修复反应,反应后用试剂盒进行纯化;(8)配制加“A”反应体系,在Thermomixer中适温反应一定时间对片段选择后的产物进行加“A”反应,反应后用试剂盒进行纯化;(9)配制P2 adapter连接反应体系,在Thermomixer中适温反应一定时间使P2 adapter与加“A”后产物连接,再使用试剂盒对连接产物进行纯化;(10)配制PCR反应体系对上一步的连接产物进行扩增,PCR产物用琼脂糖凝胶回收纯化,并溶解于适量EB solution中,至此文库制备完成;(11)构建好的文库进行质量和产量的检测,合格后用Illumina HiSeqTM平台进行测序,测序策略为Illumina PE150,周期大约为6~8周。
1.4 高通量测序分析
1.4.1 原始数据质控和过滤
Illumina HiseqTM平台测序得到的原始图像数据经过Base Calling转化为序列数据,得到最原始的测序数据文件。在Illumina HiseqTM测序数据(Raw Data)下机之后,利用Fastp软件对下机数据进行质量控制,过滤其中低质量的数据,获得高质量的数据(Clean Data)。
1.4.2 SNP检测与位点开发
利用Stacks v1.42软件进行变异检测,获得高质量的SNP位点,然后分别进行个体聚类和群体聚类,利用Stacks v1.42软件鉴别SNPs位点,再对每个个体的SNP进行统计分析,筛选出每个个体特有的SNP组合。Stacks v1.42软件的分析流程如下:
process_radtags(reads)→ustacks(stacks)→cstacks(catalog)→sstacks(allele)→populations(SNPs)。
1.4.3 群体的遗传分析
利用Stacks v1.42软件鉴定群体SNP,按照样品最低测序深度>2,样品缺失率<0.5,MAF>0.05过滤得到有效的SNP位点,进行群体分析:(1)通过RAxML软件(Stamatakis,2006)的maximum likelihood算法构建群体进化树,基于进化树分析结果,利用populations(Stacks v1.42)分析有效等位基因数目(Private)、平均观测杂合度(Ho)、平均期望杂合度(He)、核苷酸多样性(π)和平均近交系数(FIS)这6个遗传多样性指数开展遗传进化的初步分析;(2)通过ADMIXTURE(Alexander et al.,2009)软件,分析样品的群体结构,通过假设分群数(K值)(2-19),进行聚类,最后根据CV error(Cross validation error)最低点对应的K值来确定最佳分群数;(3)通过GCTA软件进行主成分分析(Principal components analysis,PCA),得到样品的主成分聚类情况,进而辅助进化分析。
2 结果与分析
2.1 RAD-seq测序数据质控和过滤
RAD-seq测序reads为基因组DNA的酶切片段,其碱基含量分布检查一般用于检测有无AT、GC分离现象。鉴于序列的随机性和碱基互补配对的原则,理论上每个测序循环上的GC含量相等、AT含量相等,且在整个测序过程基本稳定不变,呈水平线。N为测序仪无法判断的碱基类型。本项目中样品B的测序碱基含量分布如图1所示,序列的起始位置与测序的引物接头相连,因此A、C、G、T在起始端会有所波动,后面会趋于稳定。模糊碱基N所占比例越低,说明未知碱基数越少,测序样本受系统AT偏好影响越小。虚线左侧为Read1的统计,虚线右侧为Read2的统计结果。结果表明,该样品的文库构建质量和测序质量均可满足后续分析。
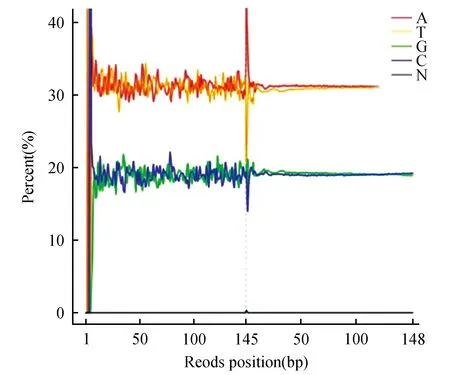
图1 样品B的碱基组成分布图 横坐标是Reads碱基坐标,坐标表示Reads上从5′到3′端依次碱基的排列;纵坐标是所有Reads在该测序位置A、C、G、T、N碱基分别占的百分比,不同碱基用不同颜色表示。Fig.1 Distribution diagram of base content The base coordinate of Reads are on the X-axis,representing the sequence of bases on Reads from 5′ to 3′; Percentages of all Reads at A,C,G,T,N bases in this sequence are on the Y-axis. Different bases are expressed in different colors.
对质量剪切后的Clean Data分别进行测序Reads数、总碱基数、GC含量和Q30比例的统计,共获得高质量的Reads数440243175,过滤后得到Clean data 129.26 G,平均每个样品5.39 G,平均GC含量为38.42%,碱基质量Q30比例达到93.67%。由于所测序列的Q30数据较高,表明碱基出错率很低,GC分布正常,数据量达到分析要求,建库测序成功。详细结果见表1。

表1 云南蓝果树基因组DNA测序数据
2.2 SNP检测与位点开发
SNP检测结果发现,所有样品酶切片段数量为341136~1282834,平均值为672131;酶切片段平均深度为12.32~70.45X,平均值为28.11X(表2),表明SNP位点检测成功。
依据材料方法部分给出的SNP检测标准,我们对样本进行了过滤,在经过填补和质控后,共计得到98498个SNP位点,每个样品检测到2029~12470个SNP位点,平均获得4104个SNP位点(表3)。根据样品最低测序深度>2,样品缺失率<0.5、次要基因型频率(MAF)>0.05的选择标准,从这些SNP中筛选出6309个有效SNP,用于群体遗传分析。样本中SNP位点的杂合率为31.32%~81.13%,平均杂合率为63.87%;样本中SNP位点的纯合率为18.87%~68.68%,平均纯合率为36.13%。由此可以看出,云南蓝果树的24个样本间SNP杂合率存在较大差异,表明它们的基因组高度杂合。另外,每个样品的转换与颠换的比值(Ti/Tv)为1.74~2.40,平均值为2.02。云南蓝果树转换与颠换的比值均大于1,说明云南蓝果树的变异更多的是发生在嘌呤与嘌呤或者嘧啶与嘧啶之间的转换。

表2 云南蓝果树的SNP检测结果
2.3 遗传分析
基于过滤得到的6 309个SNP标记,对24个云南蓝果树植株样本完成了系统进化树、群体结构和主成分分析,从基因组水平揭示了群体的遗传结构和遗传谱系。

表3 云南蓝果树的SNP信息统计

图2 云南蓝果树的系统进化树Fig.2 Phylogenetic tree of N.yunnanensis
2.3.1 系统进化树分析
基于开发出的SNP位点,采用最大似然法对24个样本构建了进化树(图2)。从进化树可以看出,24株云南蓝果树被聚成了3大类。第Ⅰ类包括16株,占总株数的66.7%;第Ⅱ类包括6株,占总株数的25%;第Ⅲ类包括2株,占总株数的8.3%。经计算,这24株云南蓝果树的Private为1 578,Ho为0.309 4,He为0.321 2,π为0.334 9,FIS为0.096 0。表4表明,系统进化树划分的3大类的Private为21~578,平均值为275.33;Ho为0.177 3~0.277 5,平均值为0.220 2;He为0.126 4~0.335 6,平均值为0.243 1;π为0.224 6~0.346 3,平均值为0.298 7;FIS为0.028 3~0.285 2,平均值为0.182 3,意味着每个分类之间的近交程度较低。其中,第Ⅰ类中表征遗传多样性的参数值都高于其他两类(Ⅱ和Ⅲ),其遗传多样性更丰富,应列为重点保护对象。

表4 云南蓝果树不同分类的遗传多样性分析

图3 云南蓝果树样品的structure结构图Fig.3 Individual cluster values(K) of N.yunnanensis with structure analysis

图4 不同K值所对应的交叉验证错误率Fig.4 The admixture validation error rate corresponding to the different K values

图5 云南蓝果树的PCA聚类图Fig.5 Principal coordinates analysis(PCA) plot generated by GCTA of N.yunnanensis
2.3.2 群体结构分析
基于开发出的6309个SNP标记,通过ADMIXTURE(Alexander et al.,2009)软件,分析样品的群体结构,分别假设分群数(K值)为2~19,进行聚类(图3)。根据聚类结果,把拥有最低交叉验证错误率(CVerror)的分群数定义为最优分群(图4)。结果显示,分群数为19时,CVerror值最小,表明样本之间遗传差异比较大,亲缘关系较远。但其曲线一直呈现下降趋势,没有最低值,不能说明k=19是最佳分组,可能所有样本的基因都是混合的,祖先也是混合的。
2.3.3 主成分分析
基于个体间SNP的差异情况,对24个云南蓝果树样本进行主成分聚类分析(图5)。结果显示,PCA分析也没有产生任何明显的分组,这与群体结构分析的结果一致(图3)。这些结果进一步支持了云南蓝果树现存植株之间遗传多样性差异较大的假设。
3 讨论
基于二代测序的简化基因组测序技术目前已作为主流方法应用于很多物种SNP标记的开发[21~24]。云南蓝果树作为云南特有的极小种群植物,曾采用SSR标记和ISSR标记研究其遗传多样性水平[25~26],但是这些标记存在通量小、准确性较低、耗时耗力、成本高等局限性[27~28],而且难以筛选出多态性足够高的遗传标记。而基于RAD-seq技术开发的SNP标记作为现在分子遗传学中最重要的分子标记,是反映生物内DNA序列变异程度的重要参数,并经统计分析可获得核苷酸多样性信息[29],具有数量多且分布丰富代表性高、遗传稳定性好、检测快速、不受基因组序列的限制等特点[22,30~31]。目前,云南蓝果树尚未获得足够的核酸序列数据,甚至也没有蓝果树科内近缘物种的可参考基因组序列,RAD-seq是一种比较理想的研究技术。从本试验所测得24个个体的有效reads数看,数据量基本均匀,未出现个体数据量差异极大的现象,基本满足后续分析的数据量要求。经过预试验,我们选择EcoRⅠ进行限制性酶切。对云南蓝果树进行基因组测序分析,共得到129.26 G的Clean data,Q30比例达到93.67%。由于所测序列的Q30数据较高,表明碱基出错率很低,GC分布正常,表明数据量达到预期目标,文库构建质量较好,测序成功,可以进行后续分析。本次研究中我们共获得SNP位点53 120个,通过样品最低测序深度>2,样品缺失率<0.5、次要基因型频率(MAF)>0.05筛选以后,得到有效SNP位点6 309。因此,本研究尝试应用简化基因组技术RAD-seq对云南蓝果树开发SNP位点,取得了成功,为云南蓝果树的群体遗传分析奠定了基础。
评估一个物种的遗传变异程度是遗传资源保护的基础和重要内容[32]。基于SNP位点的核苷酸多样性(π)、观测杂合度(Ho)、期望杂合度(He)和近交系数等参数常被用来表征群体的遗传多样性大小[24]。核苷酸多样性(π)是反映生物体内基因组DNA序列变异程度的重要参数,通过对基因组DNA的测序开发SNP位点,经过计算获得核苷酸多样性信息[29]。尤其对于双等位基因SNP位点来说,π值可以全面地测定一个群体的遗传多样性[33~35]。由于进化速率的不同,同一个物种中不同的DNA片段也可能具有不同的π值[36]。Shi等人选择了36个单拷贝核基因来推断人参属的系统发育关系,并评估同一直系同源物在二倍体和四倍体物种中是否表现出异质进化率[37]。目前通过对SNP的分析发现,很多植物的π值都低于0.1[24,29]。本研究基于开发出的SNP位点,测得24株云南蓝果树的核苷酸多样性(π)、平均观测杂合度(Ho)和期望杂合度(He)分别为0.334 9、0.309 4和0.321 2。系统进化树划分为3大类,这3大类的π值为0.224 6~0.346 3,平均为0.298 7;平均观测杂合度(Ho)为0.177 3~0.277 5,平均值为0.220 2;平均期望杂合度(He)为0.126 4~0.335 6,平均值为0.243 1。综上所述,云南蓝果树现存植株不管是整体还是各个分支的π、Ho和He值均较高,意味着云南蓝果树现存植株在SNP水平上的遗传多样性较丰富,保护其遗传资源具有重要意义。为了检验每个群体内是否存在隐藏的种群结构,近交系数(FIS)被用来衡量群体内杂合子的存在情况[33,38~39]。云南蓝果树3个分类群体的FIS为0.028 3~0.285 2,平均值为0.182 3,较高的近交水平暗示了云南蓝果树可能存在近交衰退的风险,进一步表明云南蓝果树早期幼苗在旱季全部死亡的原因,除了气候变化加剧了土壤水分短缺和自毒效应等环境因素外,也可能是由于近交衰退导致云南蓝果树适应能力较差的结果。因此,下一步工作急需通过开发云南蓝果树亲子鉴定遗传分析技术,确定种群内外植株间的亲缘关系,降低近交衰退的风险。
云南蓝果树的群体遗传分析可为评价和指导云南蓝果树拯救保护工程中遗传资源的保护提供理论依据。为了保证新建种群个体间的遗传距离保持最大,遗传资源保护最丰富,就应构建一个包括所有有效样本的样本集。由系统发育树可知,24株云南蓝果树被分为3大类,其中,M和L之间、I和H之间、U和Q之间、T和R之间、J和K之间、G和C之间、W和N之间都分别被认为遗传关系较近。因此,24株云南蓝果数最终被聚成了16个有效分支1~16(Ⅰ分类中具有11个有效分支;Ⅱ分类具有4个有效分支;Ⅲ分类只有1个有效分支)。从每个有效分支中选取1个样本作为有效样本,共得到16个有效样本。无论是对现存天然种群进行恢复,还是通过近地和迁地保护重建种群,都应构建一个包括所有有效样本1个备份以上的样本集,并且样本集里所属每个分类的遗传多样性保证为系统进化树中每个分类遗传多样性理论值的90%以上,使得遗传资源保护科学化;综合考虑环境压力和资金投入因素,可最终确定每个有效样本的备份数及恢复重建种群的规模。在野外恢复或重建种群时,确保相邻或相近的植株属于不同有效样本,并结合现存种群密度(特别是不同有效样本间的距离),使新建种群个体间的遗传距离保持最大。结合恢复或重建种群规模及其个体间的空间布局,通过ArcGIS在卫星影像图上模拟,确定保护小区面积、重建种群所需生境面积。
猜你喜欢
小猕猴智力画刊(2023年4期)2023-10-10 10:00:33
区域治理(2022年40期)2022-11-27 04:01:54
今日农业(2022年1期)2022-11-16 21:20:05
今日农业(2021年11期)2021-08-13 08:53:24
今日农业(2020年23期)2020-12-15 03:48:26
动漫界·幼教365(小班)(2019年10期)2019-10-28 02:04:20
动漫界·幼教365(大班)(2019年10期)2019-10-28 01:54:09
动漫界·幼教365(中班)(2019年10期)2019-10-28 01:53:17
农家科技中旬版(2016年12期)2016-04-16 03:41:25
遗传(2014年3期)2014-02-28 20:58:49
