
外源性一氧化碳释放分子2对脓毒症大鼠T淋巴细胞凋亡的影响
2019-11-05 00:35李佳红费东生杨松林赵鸣雁
医学研究杂志 2019年9期
李佳红 费东生 于 未 杨松林 赵鸣雁
脓毒症的主要病理生理学变化是严重全身感染引起的炎性反应过度激活、免疫功能紊乱及凝血机能障碍[1]。免疫功能紊乱尤其是免疫功能抑制被认为是脓毒症发生、发展的重要环节,而且贯穿脓毒症始终。其中免疫细胞凋亡是脓毒症免疫抑制状态发生、发展的重要机制之一,并直接影响脓毒症患者的预后和转归[2]。脓毒症可显著降低B细胞、CD4+T细胞和滤泡树突状细胞的水平,分别导致抗体生成减少、巨噬细胞活化降低和抗原递呈下降。淋巴细胞的消失尤为重要,因为它们的缺失使机体发生感染概率增加。基于对炎性反应和脓毒症认识的变化,免疫调理治疗看来是解决脓毒症的根本途径,但无论赖以指导治疗的免疫学指标还是纠正免疫紊乱的手段目前都还处在起步阶段,而且十分有限[3]。Hotchkiss等研究发现,脓毒症能够加速淋巴细胞凋亡并耗竭脾T淋巴细胞,免疫细胞凋亡与凋亡蛋白(Fas、caspase-9等)的表达有关,通过表达脓毒症大鼠模型凋亡蛋白基因,可以减少脓毒症诱导的淋巴细胞凋亡。由此,调控免疫细胞的凋亡过程有可能改善脓毒症模型的免疫抑制状态,改善预后[4]。
CO作为第二种气体信号分子,在脓毒症时能介导产生一系列细胞保护作用,在抗炎、抗凋亡、抗氧化过程中扮演着重要的角色。很多研究都证明CO的吸入提高了由盲肠结扎和穿刺(CLP)、内毒素(LPS)、粪肠球菌或大肠杆菌等诱导的败血症啮齿动物的存活率。在脓毒症小鼠中CO的供体化合物即外源性CO释放分子可以发挥类似的保护作用。值得注意的是,CO对脓毒症的抑制作用不仅限于啮齿动物模型,也包括在大型动物模型中,例如猪和非人类的LPS或肺炎链球菌的灵长类动物模型[5]。且现有研究证明HO-1和外源性CO对脓毒症中细胞凋亡有影响和作用,通过观察HO-1/CO系统干预MAPKS信号转导通路活化对免疫细胞凋亡的影响,从基因-蛋白-功能方面揭示HO-1/CO系统对于脓毒症免疫抑制调节机制,对于脓毒症治疗的探索及预防MODS的发生将具有重要的理论意义和临床应用前景[6]。CO通过调控丝裂原活化蛋白激酶(MAPKs)通路参与细胞生长、增殖、分化及凋亡等重要的生物学过程,其中ERK信号通路是经典的MAPKs途径,主要调控细胞增殖与分化;而JNK和p38通路在多种应激因子作用下均可激活,发挥介导细胞炎症、凋亡等多种生物效应,p38通路下游的凋亡蛋白(Fas、caspase-9、caspase-3等)参与淋巴细胞的凋亡,这为本研究提供了一定的理论基础[7]。本研究将应用LPS处理大鼠建立体内脓毒症炎性反应模型,观察外源性CO对LPS处理后各脓毒症大鼠模型T淋巴细胞凋亡与凋亡蛋白(Fas、caspase-9、caspase-3)的表达的关系,观察外源性一氧化碳对脓毒症模型凋亡蛋白(Fas、caspase-9、caspase-3)的表达的影响,进而探讨外源性一氧化碳释放分子2对脓毒症模型T淋巴细胞凋亡的影响。
材料与方法
1.实验动物:雄性Wistar大鼠28只,体重250~300g,由笔者医院动物实验中心提供,人工控制条件下进行饲养,室内温度为22℃,每日光照时间为12h(7:00~19:00),取食和饮水无限制。
2.药物、试剂与仪器:脂多糖(LPS)购自美国Sigma公司;CORM-2购自美国BD公司;APC Mouse Anti-Rat CD4、PE Mouse Anti-Rat CD8a Clone OX-8、 FIFC Mouse Anti-Rat CD3 Clone G4.18购自美国BD公司;Fas、caspase-3、caspase-9及小鼠β-actin单克隆抗体购自英国Abcam公司;辣根过氧化酶标记的山羊抗兔、小鼠抗小鼠抗体均购自北京中杉金桥生物技术有限公司;流式细胞仪来自笔者医院中心实验室。
3.药物的配置:CORM-2的制备,用500μl DMSO溶解 10.25mg CORM-2配制成 CORM-DMSO溶液,然后将其溶解于等体积等渗生理盐水中[8]。iCORM-2(无活性的CORM-2)溶液的制备,用DMSO溶剂充分溶解CORM-2,室温下放置2天,再通入氮气去除溶液内残余的CO。LPS溶液的制备,将10mg LPS粉末溶于10ml生理盐水,溶液浓度10mg/ml。
4.动物分组、模型制备及给药:采用数字表法将大鼠随机分为4组,即阴性对照组、LPS组、LPS+CORM-2组、LPS+iCORM-2组。腹腔注射戊巴比妥钠(50mg/kg)麻醉大鼠,经腹腔注射LPS建立脓毒症模型,经腹腔给予配制的CORM-2与iCORM-2 6mg/kg建立LPS+CORM-2与LPS+iCORM-2模型,阴性对照组:不进行任何处理;LPS组:经大鼠腹腔注射LPS,剂量为10mg/kg;LPS+CORM-2组经腹腔注射LPS,剂量为10mg/kg,待0.5h后经腹腔注射给予配置的CORM-2 6mg/kg;LPS+iCORM-2组经腹腔注射LPS,剂量为10mg/kg,待0.5h后经腹腔给予配制的iCORM-2 6mg/kg,经相应试剂预处理24h后终止实验,留取所需实验样本。
5.实验过程:给予相应试剂预处理,在24h后,收集血液、脾脏、小肠等标本,观察大鼠小肠光镜下结果进行模型验证;将抗凝血进行流式细胞检测;将部分脾脏4%多聚甲醛固定,石蜡包埋切片,HE染色,观察病理改变;其余脾脏组织置于-80℃冰箱备用做Western blot法检测蛋白的表达。
6.观察和检测指标:(1)观察大鼠小肠光镜下结果进行模型验证。(2)外周血T细胞亚群检测:在流式细胞仪上用射门法观察和计数CD4与CD8标记阳性T细胞数,计算CD4/CD8阳性细胞比值(CD4+/CD8+),从大鼠的血标本 100μl抗凝血,分别加入1μg荧光标记的单克隆抗体 CD4-APC/CD8-PE/CD3-FIFC混匀,室温避光孵育15~30min,加入2ml 1×RBC Lysis Buffer,摇匀后室温避光孵育15min,300×g离心5min,弃上清,PBS 500μl悬浮,送笔者医院中心实验室用流式细胞仪检测CD3+CD4+、CD3+CD8+的绝对值及CD4+/CD8+的比值。(3)取脾脏病理学观察,取脾脏组织 10%甲醛固定,常规石蜡切片,HE染色,普通光镜下观察脾脏组织的情况。(4)Western blot法检测Fas、caspase-3、caspase-9蛋白的表达:BCA法测蛋白浓度,取25μg蛋白进行不连续聚丙烯酰胺凝胶电泳(SDS-PAGE),电转移至聚偏二氟乙烯膜(PVDF),用含5%脱脂奶粉的TBST室温封闭1h,加入相应一抗,4℃孵育过夜,洗膜,加入相应的辣根过氧化酶标记的二抗,室温孵育1h,ECL化学发光法检测,以β肌动蛋白作为内参。

结 果
1.模型的验证:与阴性对照组大鼠比较,LPS组大鼠小肠明显充血水肿伴大量炎性细胞浸润,24h后脓毒症模型造模成功(图1、图2)。
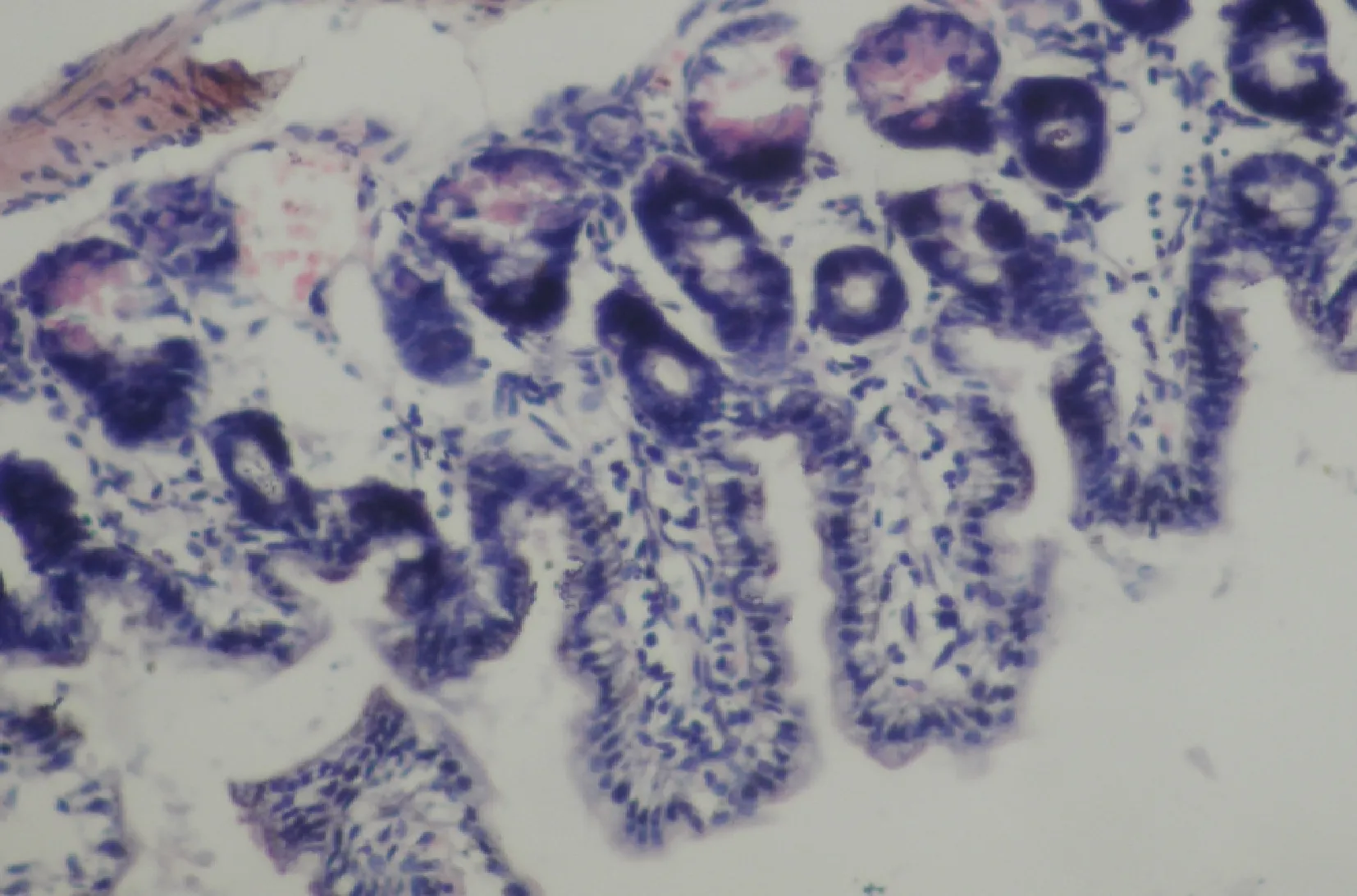
图1 阴性对照组大鼠24h后小肠光镜下病理形态结果小肠绒毛排列有序,尖耸致密,肠腔内极少量炎性渗出以及中性粒细胞浸润,形态正常(HE染色,×200)

图2 LPS组大鼠24h后小肠光镜下病理形态结果小肠绒毛变短,排列基本整齐,但充血、水肿明显,可见炎性细胞浸润,管腔内微血栓形成(HE染色,×200)
2.脓毒症患者T淋巴细胞亚群变化:分析脓毒症患者 T 淋巴细胞各亚群构成比的变化,与阴性对照组比较,LPS组CD4+与CD8+T淋巴细胞数明显减少(P<0.05);与阴性对照组比较,LPS组CD4+/CD8+比值明显减少,差异有统计学意义(P<0.05)。与LPS组比较,LPS+CORM-2组CD4+与CD8+T淋巴细胞数明显增加(P<0.05);与LPS组比较,LPS+CORM-2组CD4+/CD8+比值明显增加,差异有统计学意义(P<0.05);与LPS组比较,LPS+iCORM-2组CD4+与CD8+T淋巴细胞数比较差异无统计学意义;与LPS组比较,LPS+iCORM-2组CD4+/CD8+比值比较差异无统计学意义(表1),流式细胞仪检测结果(图3)。

表1 脓毒症患者 T 淋巴细胞亚群构成比的变化
与阴性对照组比较,*P<0.05;与LPS组比较,#P<0.05

图3 流式细胞仪检测脓毒症大鼠阴性对照组、LPS组、LPS+CORM-2组、LPS+iCORM-2组CD4+T淋巴细胞、CD8+T淋巴细胞凋亡及两者的比值
3.病理形态学观察:阴性对照组大鼠脾脏红、白髓结构清晰,小梁结构明显,白髓内含有大量淋巴小结;LPS组与LPS+iCORM-2组脾脏红、白髓分界尚清,脾白髓淋巴细胞减少,脾小结体积变小;LPS+CORM-2组较LPS组脾索增宽,其内淋巴细胞数量增多,同时淋巴结淋巴滤泡生发中心增大(图4)。与阴性对照组比较,LPS组T淋巴细胞明显减少;与LPS组比较,LPS+CORM-2组T淋巴细胞明显增加,阴性对照组与LPS+iCORM-2组比较,T淋巴细胞数比较差异无统计学意义(图5)。

图4 各组大鼠HE染色脾脏组织病理切片A.阴性对照组(×40);B.LPS组(×200);C.LPS+CORM-2组(×200);D.LPS+iCORM-2组(×200)
4.外源性一氧化碳2对脓毒症模型凋亡蛋白Fas/Fasl、caspase-9、caspase-3的表达的影响。与阴性对照组比较,LPS组与LPS+iCORM-2组的Fas、caspase-9和caspase-3的表达明显增加,差异有统计学意义(P<0.05);与LPS组比较,LPS+CORM-2组Fas、caspase-9和caspase-3的表达明显减少,差异有统计学意义(P<0.05);与LPS组比较,LPS+iCORM-2组的Fas、caspase-9和caspase-3的表达差异无统计学意义(图6)。

图5 各组T淋巴细胞相对百分数与阴性对照组比较,LPS组T淋巴细胞明显减少,与LPS组比较,LPS+CORM-2组T淋巴细胞明显增加
讨 论
免疫功能紊乱尤其是免疫功能抑制被认为是脓毒症发生、发展的重要环节,而且贯穿脓毒症始终。Hotchkiss等[10]研究显示脓毒症患者脾脏大量的淋巴细胞凋亡,且在最近的脓毒症动物模型以及死于脓毒症和多器官衰竭的患者中,已经证明了脓毒症通过细胞凋亡诱导淋巴细胞大量丢失[9~12]。在目前的研究中,脓毒症中淋巴细胞的凋亡可能是削弱免疫系统的原因,进而导致了炎性反应爆发[11,13]。脓毒症主要诱导各种淋巴细胞亚群,即B细胞、CD4T细胞、CD8T细胞,研究了脾脏CD4T细胞、CD8T细胞的凋亡情况[14]。本研究发现,脓毒症大鼠24h后与阴性对照组比较,脾脏淋巴细胞明显减少,即出现了免疫抑制,导致炎症的爆发;经外源性一氧化碳释放分子2干预后,脓毒症大鼠脾脏淋巴细胞明显增加,外源性一氧化碳释放因子2可削弱免疫抑制作用,进而发挥抗炎作用。

图6 Western blot法检测脓毒症大鼠各凋亡蛋白的表达与阴性对照组比较,*P<0.05;与LPS组比较,#P<0.05
HO体系是内源性CO产生的主要体系。HO-1被诱导后其表达增加的幅度可高达100倍,具有抗氧、抗炎、抑制血小板凝集、细胞保护、调节血管张力等重要作用,在维持组织和细胞功能以及内环境的稳定上有着广泛的生理作用[15]。有研究提示通过CORM释放的外源性CO能够缓解内毒素型和多重感染型脓毒症的急性炎症。相关研究使用LPS或者盲肠结扎穿孔模型(CLP),CORMs输注能够减少这些类型脓毒症模型的病死率。输注CORM-2能够增加野生小鼠的噬菌作用,在CLP后24h输注能在Hmox1-/-小鼠脓毒症模型中起到保护作用[16]。CO亦可减少促凋亡蛋白p53表达和线粒体释放细胞色素C抑制凋亡,故CO可作为凋亡抑制剂。CO通过调控JNK和p38通路发挥介导细胞炎症、凋亡等多种生物效应,进而再通过Fas/Fasl途径调控淋巴细胞的凋亡[17,18]。因而了解细胞凋亡的确切途径将有助于发现治疗脓毒症的新疗法,细胞凋亡的起始有两个主要途径,分别Fas/Fas配体介导的caspase-8通路和caspase-9通路,caspase-8或caspase-9随后激活caspase-3,经共同途径最终将导致淋巴细胞的凋亡[13,18]。Fas(CD95)/Fas配体(CD95L)系统是调节细胞凋亡的一个关键。
Fas通过其配体FasL的结合可以诱导caspase-9激活并导致下游激活半胱天冬酶随后切割关键调节蛋白,调节caspase-3等导致淋巴细胞的凋亡[14,19]。本研究经此通路发现在脓毒症大鼠组(Fas、caspase-9、caspase-3)凋亡蛋白明显增加,加速了T淋巴细胞的凋亡,免疫反应受抑制,在经外源性一氧化碳释放分子2干预后,凋亡蛋白(Fas、caspase-9、caspase-3)表达明显减少,T淋巴细胞凋亡减少,增强了免疫反应,进而发挥其抗炎作用。
综上所述,脓毒症大鼠在受到CORM-2干预后,引起凋亡蛋白(Fas、caspase-9、caspase-3)表达的明显减少,说明外源性一氧化碳释放分子2能够在脓毒症模型中抑制凋亡蛋白的表达,且在CORM-2干预组的脓毒症大鼠T淋巴细胞较脓毒症组明显增加,即通过Fas/Fasl配体调控下游的(caspase-9、caspase-3)抑制T淋巴细胞的凋亡,外源性一氧化碳释放分子2通过抑制凋亡蛋白的增加而有效的削弱了脓毒症对T淋巴细胞的凋亡效应。由此可以说明外源性一氧化碳释放分子2通过抑制凋亡蛋白的表达减少T淋巴细胞的凋亡。
此外,本研究的创新点为阐述了外源性一氧化碳对脓毒症模型的免疫系统方面的作用,证实了外源性CO通过抑制凋亡蛋白的表达减少T淋巴细胞的凋亡,这也为外源性CO的抗凋亡作用提供了新的理论基础,外源性CO诱导的抗凋亡作用可能涉及其他的信号转导通路(其上游P38通路等),本研究仍有不足,需要在今后开展的研究中进一步证实。
猜你喜欢
中华养生保健(2020年4期)2020-11-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
保健与生活(2020年5期)2020-03-20
中国中医急症(2019年10期)2019-05-21
中成药(2017年9期)2017-12-19
中国兽医杂志(2016年3期)2016-06-22
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国卫生标准管理(2015年18期)2016-01-20
中国卫生标准管理(2015年15期)2016-01-15
中国当代医药(2015年16期)2015-03-01
