
晶体化学教学中单晶结构解析存在的问题
2019-11-04 00:23魏振宏陈超丁顺民
教师博览·科研版 2019年8期
关键词:晶体结构
魏振宏 陈超 丁顺民


[摘 要] 晶体是由原子、分子或离子在三维空间按一定规则呈周期性排列而成,在外形上表现为具有一定形状的几何多面体。晶体内部结构可以通过X-射线单晶衍射实验及相应的程序解析获得,但除了经验丰富的晶体学家,一般的科研工作者对单晶解析都感到束手无策。晶体化学中,要顺利解析出单晶结构,关键在于初始套的确定、无序的精修、加氢三个方面。
[关键词] 晶体化学;晶体结构;单晶解析
晶体化学是研究晶体在原子水平上的结构,揭示晶体的化学组成、结构和性能三者之间内在联系的学科。自1966年以后,由于计算机控制的自动单晶衍射仪和与之匹配的晶体结构分析软件的迅速发展和普及,X-射线单晶衍射测试成为获取分子立体结构和键参数最权威的技术手段。截至目前,剑桥晶体数据库(CCDC)已收录的无机化合物、有机化合物和金属配合物的晶体结构数量已超过100万种,并且仍以每年超过5万的数量在增加。这些大量的晶体化学结构信息为广大科研工作者深入研究反应机理、指导合成和深入探讨物质分子构型与分子化学活性间的内在联系提供了可靠的依据。
目前,国内高校和科研机构已经基本配备了X-射线单晶衍射仪,势必要求每位与之相关的研究人员都掌握X-射线单晶结构的解析方法,因此晶体化学成为高校化学专业研究生的选修课程。然而,一线科研工作者,特别是初学者,在解析晶体结构时经常感到困惑,不知从何下手。其实,我们知道解析晶体结构是一门经验技术活,教师除了需要在课堂上详细介绍教材上的晶体解析的步骤和方法外,还需要把自己多年在解析单晶时积累的经验、技巧和心得告诉学生。通过最近几年晶体化学的教学,我们发现研究生经常对初始套的解析、无序的精修和如何加氢感到困难重重,因此,我们有必要将这些问题进行归纳总结,希望利用积累的经验帮学生熟练掌握晶体解析这门技术。
一、初始套的确定
在解析结构前,我们需要评判衍射数据的好坏。经常有些同学在解析的时候从来就不关注衍射数据的好坏,拿来数据就直接进行解析,等解到一半时才发现晶体数据质量很差,根本无法得到最终的结构,或者晶体数据质量一般,虽然能够解析出大致的结构,但是最后的输出文件cif达不到论文发表的要求。因此在解析前,一定首先对数据好坏进行评估,避免做无用功。如果发现晶体质量太差,衍射强度数据太弱,我们需要重新培养单晶,收集较好的衍射数据后再进行解析。
我们可以从以下几个指标对数据好坏进行评判:数据分辨率(Resolution)要求不得低于0.84;数据的完整度(Complete)不得低于95%;数据冗余度(Redundancy)越大越好;平均信噪比(Mean I/sigma)不能低于2;等效点平均标准误差(Rint)不得大于12%;可见衍射点N(int>3sigma)占总衍射点数N(total)比例一般来说要大于50%。在上述指标中,分辨率、完整度、平均信噪比、等效点平均标准误差这四个指标在checkcif过程中会进行检查,不符合要求会报错。
数据评估合格后,接着需要确定晶体的空间群,空间群可以明确反映一种晶体可能具有的对称元素种类和这些对称元素在晶胞中的位置。一般解析程序通过计算会自动提供一系列参考选择,并给出可能的空间群及其对应的综合因子CFOM,CFOM小于1表明建议的空间群很有可能是正确,且CFOM值越小,空间群正确的概率越大。如果我们没有特别把握,应该优先按照程序自动默认的空间群进行解析;当默认的空间群不能确定晶体结构,且有其他空间群可选择时,先选CFOM最小者;如果按以上选择仍无法解出合理结果,还可尝试先将空间群的对称性降低,解出初始结构,再升高空间群。升高对称性操作步骤如下:首先,利用PLATON软件打开.res文件,选择菜单栏 (PLATON graphical menu), 点击SYMMETRY菜单栏中的“ADDSYM-SHX”;然后在DOS系统中将.res文件复制到.ins文件,再计算确定最终结构。
二、无序的精修
在单晶结构解析过程中,经常会遇到无序结构,主要原因是由于熵效应导致了晶体微观结构并不完美,许多晶体存在杂质和晶体缺陷。对无序的精修处理是最令人头痛和棘手的问题,因此我们需要了解晶体结构中产生无序的本质原因,并掌握无序基团的常规处理方法。
对于无序的精修主要任务是确定无序原子的两个不同的位置坐标及各自占有率。位置相对占有率可以自行给定,也可以自由精修得到,而找到这两个位置的坐标通常要复杂得多。SHELXL程序通过将无序原子劈成几组进行精修,无序组分的占有率被允许自由精修,与PART指令一起,自由变量的引入使得无序的精修变得容易。
晶体结构产生无序的本质原因可归纳为两大类:第一类是位置无序,它是由晶体中原子的热运动和单键的旋转造成的,使得分子以不同的构象并存于不同晶胞中;第二類是占有率无序,是由半径相近的不同元素在晶体中相互取代、相互掺杂引起的。这两类原因造成的结构无序,在单晶结构精修过程中将采取不同的处理方法:
1.位置无序的精修
位置无序描述晶体中的分子或分子的一部分统计性地分布在两个或两个以上位置,总占有率为1。这种情况有可能发生在单个晶胞中(动态无序,固体状态下的真实热运动),也可能发生在不同的晶胞中(静态无序,类似于运动)。在精修过程中,动态和静态无序都用相同的方式处理。
在静态无序中,一个分子可以有两个能量相近的构象,从空间水平看,这种无序由两种构象的叠加造成,因此可以把无序原子分裂成两部分进行解析处理。
相对于静态无序,动态无序结构更加复杂。例如,叔丁基的每个旋转角度在能量上是相似的,并且没有空间位阻,那么这组原子可以在晶体中自由旋转,从空间水平看,这组原子为旋转的圆球,因此这种无序处理起来十分困难。一般,我们将这种无序简化为每个原子占据两到三个位点,并接受原子的较大热位移参数。当然,我们可以通过收集低温数据减少或避免这种动态无序的产生。
2.占有率无序的精修
占有率无序描述的是在两个晶胞中的相同位置被半径接近的不同原子所占据,总占有率为1。这种类型的无序在矿物晶体中尤其常见,如:某些沸石中铝和硅原子共享相同的位置;在生物结构中,有时水分子与钠、氯或其他离子共用一个位置,在晶体解析中我们可以通过该位置原子的位移参数是否太大或太小的报错信号判断该位置是否存在占有率无序。
另外,位置占有率无序也可以表现为原子(或基团)在某一位置上的占有率小于1,如非配位溶剂分子通常只占据晶格中大约一半的空穴。溶剂分子异常高的位移参数是原子位置部分占据的表现。然而,我们应该考虑到,由于它们的热运动性,即使完全占据的非配位溶剂分子也往往显示出较高的位移参数。
3.如何寻找第二个位点
如果在差值傅里叶图中发现了第二组峰值,则认为有明显的无序;如果原子的热位移参数表现出强烈的各向异性,同样表明有无序的存在,这时SHELXL程序自动将此原子的两个可能的位置坐标写入.lst文件。然而,并非所有“需要被分裂”的原子都应该进行劈分,有时原子在单个位置上的各向异性运动反而是对其结构更好的表达。如果无序原子彼此相距太远,或者热位移参数的各向异性不足,SHELXL都不会建议劈分该原子,我们只能用残余电子密度峰的坐標作为第二位点。有时,我们可以对分裂原子的两个位置使用相同的初始坐标,让SHELXL在后续精修过程中给定最终的位置坐标。对特殊位置原子的精修相对容易,无序原子的第二个位置可以通过对称操作直接从第一部分原子坐标计算出来。例如,在镜面、二重轴或反转中心中,位置占有率为10.5000,三重轴时为10.3333,四倍轴时为10.2500。
4.限制性指令和强制性指令的使用
在对无序结构精修时,总会引入一些限制性指令,这会大大地增加精修参数。限制性精修可以通过对结构的观察分析,为特定原子提供目标参数值,因此,晶体学家可以依据一些已知化学和物理的结构信息通过限制指令对结构进行精修。另外,在有些无序例子中,强制性命令也会用于精修过程中。与限制性指令不同,强制性指令是通过数学方程,严格联系起两个或更多的参数,或赋予特定参数特定值,从而减少参与精修独立参数的数目。不过,强制性指令的使用会拉高反应精修结果的R值。
5.无序基团精修举例
(1)高氯酸根无序
ClO4?基团中Cl原子占据相同的位置,而氧原子具有两个不同的取向,如图1所示。在最初的差值傅里叶图中可能会清楚地显示出两组不同氧原子的位置,也可能只显示出一组氧原子,但在各向异性精修后,由于氧原子具有较大的位移参数,根据程序在. lst文件中的提示,将氧原子的位置劈分成两组。在对该无序基团进行精修的过程中,可以限制的参数包括Cl?O的键长、氧原子之间的距离(DFIX)、中心Cl原子在同一个位置上(EXYZ, EADP)、各向异性位移参数(ISOR)、两组氧原子的占有率之和为1(PART中,位移参数为21和-21)等。
(2)乙基无序
乙基这样的末端基团,很容易出现无序情况,如图2所示,在对其无序精修前可以先对其进行键长的限制,限制键长为1.54 (0.01) ?,然后再进行无序拆分操作。具体如下:在.ins文件中找到C1、C2原子,将其坐标参数相邻放置。首先,在C1之前加PART 1,C2之后写PART 2;然后,在PART 2 之后添加乙基无序原子的坐标参数;接着,在C2A原子后加PART 0;最后,将PART 1中所有原子的位置占有率改成“21.0000”,PART 2中所有原子的位置占有率改成“-21.0000”。
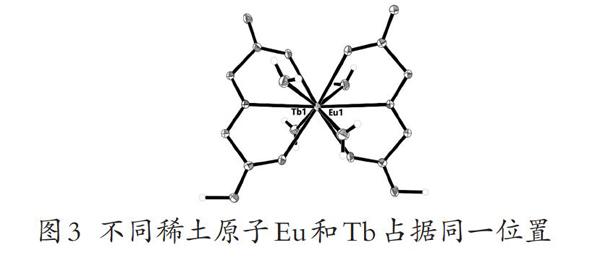
(3)同一个位置被不同原子占据
在晶体解析过程中,经常会碰到两个不同的原子出现在同一个位置,这种无序常见于矿物晶体和一些稀土离子存在的配位化合物中,例如在Tb和Eu与某有机化合物配位的晶体中,由于镧系收缩,稀土元素半径接近,因此结构中Tb和Eu占据同一位置,实际的拆分操作为:在.ins文件中添加指令EXYZ Tb1 Eu1,规定Tb1和Eu1具有相同的晶体学坐标;加指令EADP Tb1 Eu1,给两原子指定相同的各向同性或异性的位移参数;由于Tb1和Eu1在特殊位置上,占有率改为10.25,最后在FAVR后加0.5。
三、加氢
在晶体解析过程中,在确定完初结构,对非氢原子进行各向异性精修后,接下来的任务就是对原子进行加氢。加氢的方式有两种,一是理论加氢,另一种是傅里叶加氢,在这块教学时发现的问题是学生不太清楚理论加氢命令的实质及傅里叶加氢的方法,需要做详细的阐述。
所谓理论加氢是基于已知分子几何构型直接通过加氢指令进行加氢的方法。理论加氢的指令格式为:“HFIX ab 需加氢原子名称”,其中a表示氢的类型,当 a = 1、2、 3(或13)、4 ,分别表示叔氢、仲氢、伯氢、芳香氢等;b表示固定的类型,当b = 1,表示固定了它的坐标、占有率、位移,b = 2,表示固定了它的占有率、位移,b = 3,表示只固定了它的坐标。最后XL精修,检查H加得是否合理,如不合理,继续精修。通常有机骨架上的C和N以及羟基(?OH)上的H都可以利用几何加氢的方法来处理,但是水分子上的氢只能通过傅里叶加氢来完成。
在晶体学数据比较好的情况下,水分子的H原子在残余峰上还是有迹可循的。通常通过傅里叶图残余峰指认水上氢需要考虑两方面的因素:1)H?O?H的键长和键角(键长应在0.65?1.15 ?之间,键角在95?115°之间);2)一定要形成氢键,水分子的H应位于能形成合适的氢键位置上,而不是随意的位置。基于以上两点考虑,在用SHELXTL程序精修时,可以在主体骨架都确定之后把残余峰的数量改为50,甚至更大(PLAN 50),然后在O周围的残余峰中仔细辨认,把位置合适的残余峰定为H。从残余峰中得到H原子,键长一般不是理想的键长,而且位置在精修过程可能会发生改变,为了解决这些问题,我们需要限制性指令DFIX把H?O键长调整为理想的0.85 ?。这样就基本上可以得到较好的水分子的H原子了。此外还可以通过WinGX程序包中的CACL-OH来计算水上的H原子。如果以上两种方法都得不到较好的H原子,建议放弃加氢,因为数据质量不好的话,水分子上的氢很难确定出来。
总之,解析物质的结构对了解其在实际中的应用具有非常重要的意义,虽然对初学者来说该过程看起来比较复杂,但是只要熟悉指令,勤加练习,并且参考常见化合物的解析方法,结合实际,最终也会成为熟练的晶体学专家。
责任编辑 李杰杰
猜你喜欢
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
新乡学院学报(2016年6期)2016-12-01
贵州科学(2016年5期)2016-11-29
衡阳师范学院学报(2016年3期)2016-07-10
火炸药学报(2014年3期)2014-03-20
郑州大学学报(理学版)(2014年2期)2014-03-01
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年5期)2014-02-28
无机化学学报(2014年1期)2014-02-28
