
莠去津分子印迹聚合物微球的制备及其吸附性能与在食品检测中的应用
2019-10-29 06:38张凯杰秦思楠赵春娟高文惠
食品科学 2019年20期
张凯杰,秦思楠,赵春娟,高文惠*
(河北科技大学生物科学与工程学院,河北省发酵工程技术研究中心,河北 石家庄 050000)
莠去津是一种广泛用于玉米、大豆、甘蔗、高粱等农田作物的三嗪类除草剂,结构式如图1所示[1]。由于莠去津农药用量较大、性质较稳定,在土壤中不易降解,而且在水中有较高溶解度,因此在施用过程中可能对土壤、地表水及饮用水造成污染,从而严重威胁人类健康和环境安全[2-3]。研究表明,莠去津为内分泌干扰物质,对人体有潜在致癌、致畸、致突变的作用,同时对动物和水生生物的生殖发育存在毒害作用[4-5]。
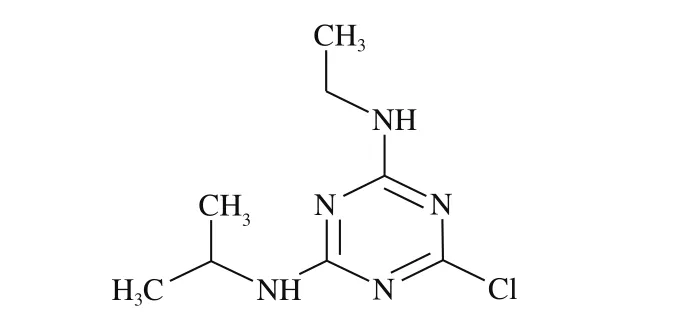
图1 莠去津结构示意图Fig. 1 Chemical structure of atrazine
目前常用于三嗪类农药的检测方法主要包括高效液相色谱(high performance liquid chromatography,HPLC)法[6-8]、气相色谱法[9-11]、液相色谱-质谱联用技术[12-14]、气相色谱-质谱联用技术[15-17]和毛细管色谱法[18-19]等。但通常情况下样品成分复杂,而三嗪类除草剂在样品中含量很低,在样品前处理过程中尽量减少基质效应对分析效果的影响是目前学者需要解决的重要问题。因此,探索一种干扰小、识别性强的分离富集方法具有特别重要的意义。
分子印迹技术(molecularly imprinted technique,MIT)是一种以目标分子为模板,合成具有特殊分子识别功能的分子印迹聚合物(molecularly imprinted polymers,MIPs)的技术[20]。因MIPs具有高选择性和特异识别的能力,且制备简单、成本低、耐苛刻条件、可重复利用等,已被应用于固相萃取[21-24]、手性分离[25]、仿生传感器[26]、色谱分析等研究领域[27]。
MIPs有多种制备方法,如本体聚合法、沉淀聚合法、悬浮聚合法、表面印迹法等,本体聚合法虽然操作简单、易控制,但在制备过程中需研磨,可能破坏聚合物内部的印迹孔穴,严重影响聚合物的选择性和吸附性,而沉淀聚合法、悬浮聚合法、表面印迹法克服了本体聚合法的缺点。关于莠去津MIPs的制备已有多篇报道[28-31],Gkementzoglou等[29]运用微乳液聚合法和沉淀聚合法制备了莠去津纳米MIPs微球,结果显示由沉淀聚合法制备的MIPs对目标分子表现出更高的吸附容量,并从聚合机理上做出了解释,沉淀聚合法制备的MIPs可在聚合物的内部和表面同时形成结合位点,而微乳液法仅在表面形成。陈凯尹等[30]以莠去津为模板分子,甲基丙烯酸(methacrylic acid,MAA)为功能单体,采用沉淀聚合法制备了粒径约210 nm的莠去津纳米MIPs微球。Liu Guangyang等[31]在甲醇中以Fe3O4-壳聚糖纳米粒子为载体,采用表面印迹法制备了一种新型的莠去津磁性MIPs,用于自来水中莠去津的吸附。而如果将MIPs作为固相萃取材料应用于样品前处理,不仅选择性强,样品净化效果好,而且还具有固相萃取材料能反复使用,能有效防止色谱柱污染等优点。
本实验以莠去津为模板分子,MAA为功能单体,采用沉淀聚合法制备分子印迹聚合微球(molecularly imprinted polymer microspheres,MIPMs),并将其应用于食品中三嗪类农药残留分析,建立食品中莠去津及其结构类似物农药残留量的分子印迹固相萃取-HPLC检测方法。
1 材料与方法
1.1 材料与试剂
莠去津(9 6%);西玛津(9 8%);莠灭净(9 5%);扑草净(9 7%);M A A(分析纯);乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA,分析纯);偶氮二异丁腈(azobisisobutyronitrile,AIBN,分析纯);甲醇、乙腈(色谱纯);冰乙酸(分析纯);实验用水为超纯水。
1.2 仪器与设备
L C-2 0 A H P L C仪 日本岛津公司;EVOLUTION220型紫外-可见分光光度计 美国Thermo公司;KH5200型超声波清洗器 昆山禾创超声仪器有限公司;ASE-12型固相萃取仪 天津奥特赛恩斯仪器有限公司;TGL-15B型高速离心机 上海安亭科学仪器厂;DZF-6320型真空干燥箱 上海新苗医疗器械制造有限公司。
1.3 方法
1.3.1 紫外光谱测定
配制1 mmol/L莠去津的乙腈标准溶液、2 mmol/L MAA的乙腈溶液。取5 支10 mL比色管,向各管中加入莠去津标液150 μL,按照模板分子与功能单体物质的量比为1∶2、1∶4、1∶6、1∶8、1∶10向比色管中加入MAA的乙腈溶液150、300、450、600、750 μL。将5 支比色管用乙腈定容至10 mL,超声10 min,于4 ℃静置14 h,对上述混合溶液进行紫外光谱扫描。
1.3.2 莠去津MIPMs的制备
取2 个150 mL圆底烧瓶,将0.2 mmol模板分子莠去津(空白不加)和0.8 mmol功能单体MAA放入圆底烧瓶中,用乙腈溶解,室温超声30 min,4 ℃预聚合6 h,形成模板分子-功能单体复合物,取出至室温后加入1.5 mL EDMA和35 mg AIBN,超声混合10 min。然后向圆底烧瓶中通N210 min除去氧气,在60 ℃恒温水浴中反应24 h;聚合完成后,将聚合混合液于10 000 r/min离心5 min,得白色印迹聚合物(MIPs)和非印迹聚合物(NIPs),用甲醇-乙酸(90∶10,V/V)混合试剂作为洗脱剂去除MIPs中的模板分子,再将MIPs微球于45 ℃,真空度为0.04 MPa条件下干燥6 h,然后放入干燥器中备用,即得到可使用的MIPMs。
1.3.3 结合量实验
1.3.3.1 不同聚合物吸附性能的考察
分别向10 mL具塞比色管中加入不同致孔剂量(15、30、50、70 mL)和不同聚合温度(50、60、70 ℃)制备的莠去津MIPMs及NIPs各20 mg,然后向各比色管中加入1 mmol/L莠去津的乙腈标准溶液5 mL,超声混匀,25 ℃恒温振荡24 h后,10 000 r/min离心5 min,上清液过膜,供HPLC分析。根据HPLC分析结果可计算MIPMs和NIPs的吸附容量(式(1))和印迹因子(imprinting factor,IF,式(2))。

式中:Q为聚合物对莠去津的吸附容量/(μmol/g);C0为莠去津的初始浓度/(μmol/L);C为莠去津的平衡浓度/(μmol/L);V为添加的溶液体积/mL;M为称取聚合物的质量/mg。

式中:QMIPMs为MIPMs对莠去津的吸附容量/(μmol/g);QNIPs为NIPs对莠去津的吸附容量/(μmol/g)。
1.3.3.2 静态吸附实验
称取20 mg莠去津MIPMs及NIPs分别放入10 mL具塞比色管中,加入一定量莠去津的乙腈标准溶液,浓度范围在0~600 μmol/L之间,将比色管置于25 ℃恒温振荡24 h后,10 000 r/min离心5 min,上清液过膜,供HPLC分析。
1.3.3.3 竞争性吸附实验
称取20 mg莠去津MIPMs放入10 mL具塞比色管中,然后加入5 mL一定浓度的西玛津、莠去津、莠灭净和扑草净的乙腈溶液,混合物在25 ℃恒温振荡24 h后10 000 r/min离心5 min,上清液过膜,供HPLC分析。
1.3.4 莠去津-MISPE柱的制备
称取适量MIPMs,放入水中进行超声分散,然后将匀浆全部装入具砂芯玻璃层析柱内,并装入固相萃取装置中待以后实验备用。柱内聚合物的高度2 cm。填装完成后,在柱顶端塞入脱脂棉,轻压脱脂棉使其更加紧实。
1.3.5 样品处理
1.3.5.1 提取
分别称取磨碎或切碎后样品5.000 g,果蔬样品中加入3.000 g无水硫酸钠,除去过多的水分,谷物中不加,用20 mL乙腈进行提取(分两次,每次10 mL),搅拌均匀,超声提取20 min,在4 000 r/min离心10 min,合并上清液,过滤,用乙腈定容至20 mL,待过固相萃取柱。
1.3.5.2 提取液净化
1)活化:先用10 mL水浸润柱子,再用10 mL甲醇过柱活化。2)上样:取样品提取液1 mL移入固相萃取柱,设置固相萃取仪压力为45 kPa。3)淋洗:当样品提取液全部过柱后,用20 mL水淋洗固相萃取柱。4)洗脱:用10 mL甲醇-乙酸(90∶10,V/V)溶液过柱洗脱。收集洗脱液,定容至10 mL,过0.45 μm的微孔滤膜,供HPLC分析[32]。
1.3.6 HPLC测定条件
色谱柱:Symmetry®C18(4.6 mm×250 mm,5 μm);检测波长:220 nm;流动相为甲醇-水(86∶14,V/V);流速1 mL/min;柱温30 ℃;进样量20 μL。
1.4 数据处理与分析
采用Origin 8.0和Excel 2007对实验数据进行作图及统计分析,每组实验重复测量3 次,数据均为3 次平行实验平均值。
2 结果与分析
2.1 模板物质与功能单体最佳比例的选择
合成MIPs时,功能单体用量直接影响印迹聚合物对模板分子的吸附能力。功能单体加入量少时,模板分子与功能单体在预聚合过程中形成的复合物不稳定,特异性识别能力不足;当功能单体用量过多时,会导致聚合物的非特异性吸附能力增强,致使特异性识别能力变差[33]。
固定莠去津的浓度,逐渐增加MAA的浓度,用紫外分光光度计测得混合液的吸收曲线,如图2a所示。当莠去津与MAA的比例由1∶2变化到1∶10时,混合液的最大吸收波长从205 nm增加到212 nm(图2b)。当混合物体系最大吸收波长对应的吸光度变化趋于平缓时,说明混合物物质的量比例对模板分子与功能单体之间的作用趋于稳定,此时平缓区的转折点所对应的混合物比例是最佳配比[34]。由图2b可知,莠去津与MAA物质的量比在1∶4~1∶10范围之间混合物的吸光度随波长红移量的变化趋于平缓,由此推断,莠去津与功能单体MAA的最佳物质的量比为1∶4。
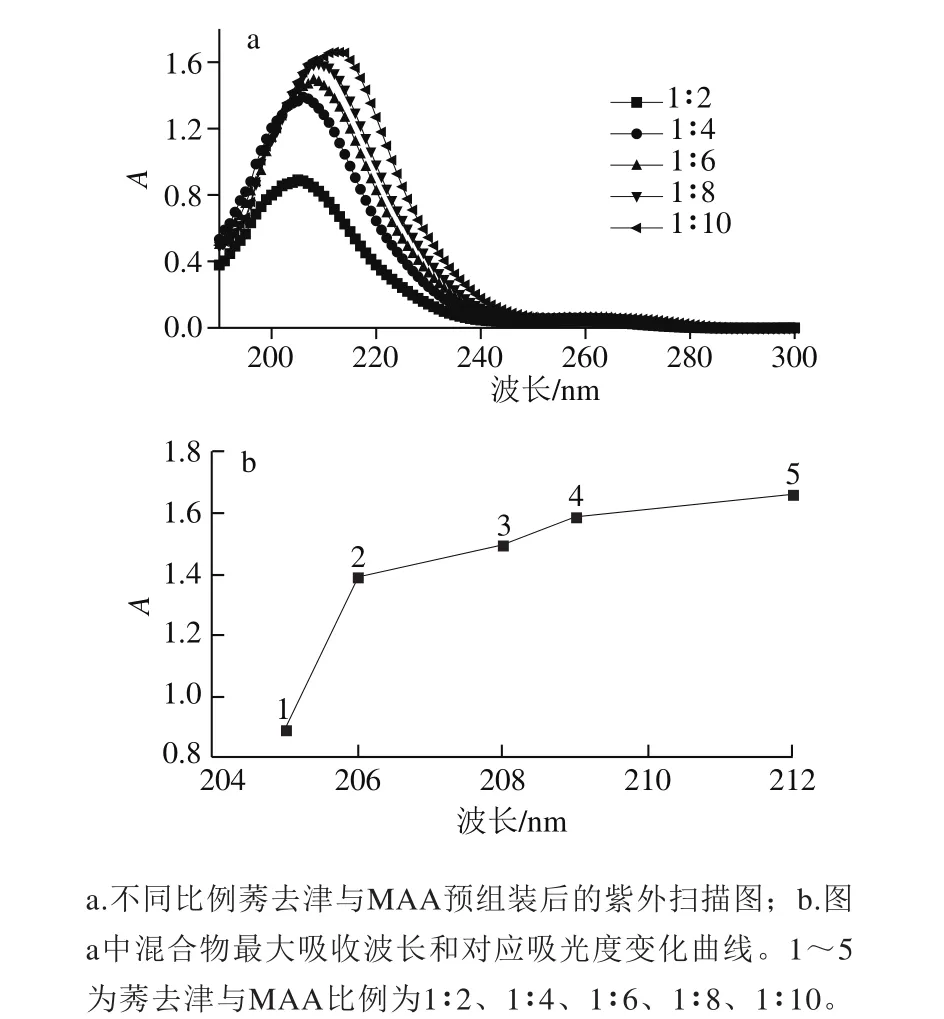
图2 莠去津和MAA比例对预组装体系紫外光谱的影响Fig. 2 Effect of different ratios of MAA to atrazine on UV spectrum of pre-assembled systems
2.2 致孔剂用量和聚合温度对聚合物吸附性能的影响
在莠去津、MAA和EDMA加入量分别为0.2 mmol、0.8 mmol和1.5 mL的条件下,改变乙腈的加入量和聚合温度,考察这些条件制备的聚合物对莠去津吸附性能的影响。通过平衡结合实验,比较各MIPMs和NIPs的吸附容量Q和IF,其中IF是评价印迹成功与否的最直观指标,结果见表1。
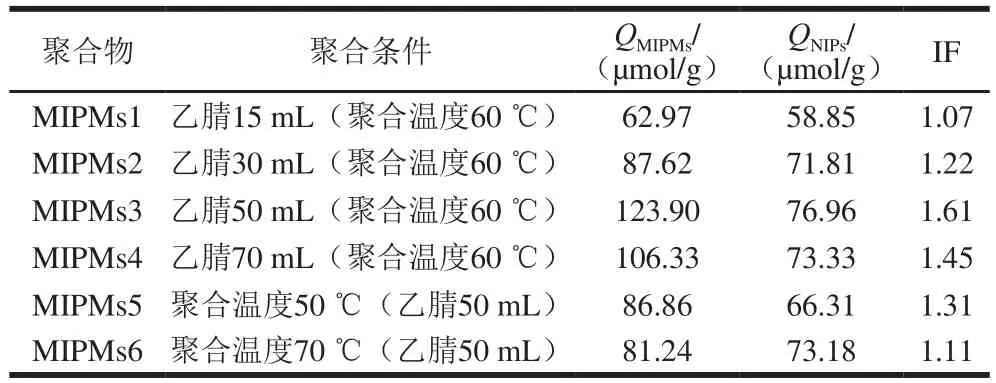
表1 不同聚合条件对聚合物吸附性能的影响Table 1 Effect of different polymerization conditions on adsorption properties of polymers
从表1可见,乙腈50 mL、聚合温度60 ℃时吸附量大而且印迹效果好。MIPMs1~MIPMs3的比较发现,增加致孔剂的用量可以提高聚合物吸附量和选择性;由图3可以看出,当乙腈用量为15 mL或30 mL时,聚合物不能形成均匀的颗粒,团聚情况比较严重,这可能是因为当致孔剂用量较少时,聚合反应接近于本体聚合;当乙腈用量为50 mL或70 mL时,沉淀聚合形成聚合微球,且粒径均匀,无团聚现象,其粒径大小分别为120 nm和200 nm左右。乙腈用量为50 mL时,聚合物微球粒径小、比表面积大,吸附量大;但当乙腈用量达到70 mL时,由于自由基单体浓度降低导致聚合反应速率减慢,形成的聚合物较少,宏观表现为材料结构松散、易碎。因此本实验选择乙腈用量为50 mL,该条件下制备的聚合物微球粒度较文献[30]小很多,且分散性好。对MIPMs3、MIPMs5、MIPMs6进行比较,研究聚合温度对聚合物微球性能的影响。结果表明,聚合温度越高,聚合速率越快,当聚合温度达到70 ℃时,出现爆聚现象,即短时间内合成大量的MIPMs,由表1可以看出,爆聚现象影响了MIPMs识别位点的形成,导致MIPMs的吸附性能下降;聚合温度为60 ℃时,12 h左右出现大量MIPMs;50 ℃时,常常出现无聚合现象,这可能是由于引发剂AIBN需要一定的温度才能进行热引发,若温度过低,无法达到引发剂的热分解温度,聚合将不能进行。综上,选用60 ℃作为最佳聚合温度。

图3 不同乙腈用量聚合物微球的扫描电镜图Fig. 3 Scanning electron micrographs of polymer microspheres with different amounts of different acetonitrile
2.3 聚合物吸附性能
2.3.1 静态平衡结合实验及Scatchard分析
一般通过静态平衡吸附法评价MIPs对目标分子的选择性能。由图4可以看出,MIPMs比NIPs对模板分子有更好的吸附性,在莠去津浓度大于300 μmol/L时,在相同浓度下,MIPMs吸附量远大于NIPs吸附量。这说明,MIPMs在制备过程中,形成了与模板分子互补的三维空穴结构,它对模板分子有记忆功能,具有特异选择性。但是合成NIPs时由于未加入模板物质,因此NIPs对模板分子无特异选择性。

图4 MIPMs和NIPs对莠去津的静态吸附等温线Fig. 4 Static adsorption isotherms of MIPMs and NIPs toward atrazine
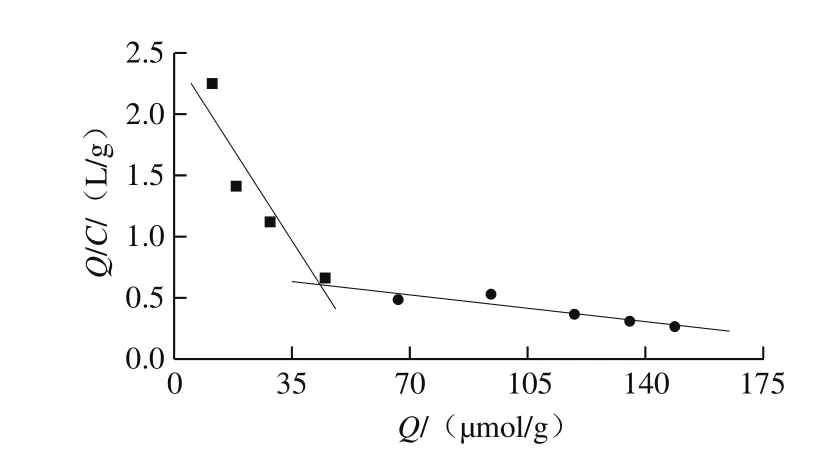
图5 MIPMs吸附性的Scatchard曲线Fig. 5 Scatchard curve of MIPMs adsorption
由Scatchard分析可以获得聚合物对目标分子吸附位点的种类、解离常数及最大吸附量等信息。Scatchard方程见式(3):

式中:Q为聚合物对莠去津的吸附量/(μmol/g);C为莠去津的平衡浓度/(μmol/L);Qmax为吸附位点的最大表观结合量/(μmol/g);Kd为吸附位点的解离平衡常数/(μmol/L)。
由图5可知,Q/C对Q是非线性的,说明MIPMs对莠去津的吸附是非均一的,原因可能是聚合过程中加热容易造成体系中热力学条件的不均匀,使模板分子和功能单体产生不同的作用形式,同时自由基聚合过程的不稳定性致使空穴的大小产生差异,影响了结合动力学特性的均一性[35]。从图5可以看出,Scatchard分析图中有两个线性较好的部分,说明MIPMs对模板分子主要存在两类吸附位点。对两个线性部分进行拟合,得到两个线性方程:第1类吸附位点的方程为y=2.486-0.042 7x,其解离常数Kd1为23.42 μmol/L,最大表观结合量Qmax1为58.22 μmol/g;第2类吸附位点的方程为y=0.735-0.002 6x,解离常数Kd2为384.62 μmol/L,最大表观结合量Qmax2为282.69 μmol/g。
2.3.2 竞争性吸附性能
为考察MIPs的特异性吸附能力,用莠去津MIPMs对莠去津及其结构类似物西玛津、莠灭净、扑草净的吸附性能进行测试。
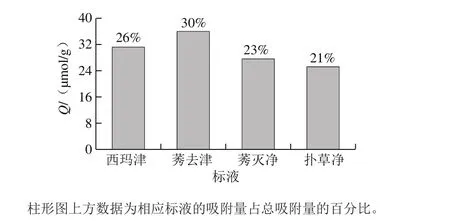
图6 MIPMs对莠去津及其结构类似物的吸附选择性Fig. 6 Adsorption selectivity of MIPMs toward atrazine and its structural analogues
由图6可知,MIPMs对4 种三嗪类除草剂均有吸附,吸附效果依次为莠去津>西玛津>莠灭净>扑草净,说明多种物质存在时MIPMs对其吸附性能存在差异,主要原因是4 种农药的结构与分子质量的大小不同。首先是MIPMs对模板物质莠去津的吸附性能最好,其次是分子质量最小的西玛津,而分子质量大于模板物质的莠灭净与扑草净吸附量较小。
2.4 分子印迹固相萃取柱的重复性和稳定性
为验证分子印迹固相萃取柱的重复性和稳定性,实验取浓度为1 mmol/L莠去津标液1 mL按1.3.5.2节方法过分子印迹固相萃取柱。由表2可以看出,使用分子印迹固相萃取柱10、30、50、70、90、100 次,莠去津过MISPE洗脱液峰面积没有发生明显变化,即固相萃取柱并未随着使用次数的增加其吸附效果有所降低,说明分子印迹固相萃取柱具有良好的重复性和稳定性。
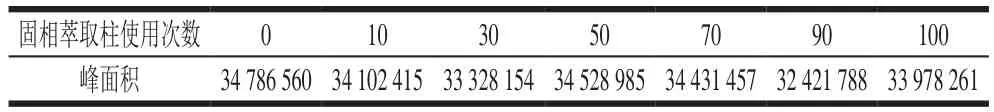
表2 固相萃取柱可反复使用次数Table 2 Reusability of MISPE column
2.5 色谱条件的选择
2.5.1 检测波长的选择
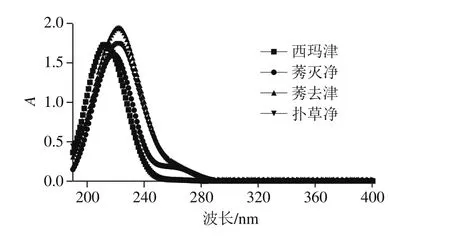
图7 4 种三嗪类农药的紫外光谱图Fig. 7 UV absorption spectra of four triazine herbicides
由图7可知,西玛津、莠灭净、莠去津和扑草净的最大吸收波长分别为212、218、222、222 nm,综合考虑各物质的紫外吸收情况,检测波长以220 nm为宜。
2.5.2 流动相比例选择
本实验分别考察不同比例的甲醇-水(90∶10、86∶14、83∶17、80∶20,V/V)对4 种三嗪类农药保留行为的影响。流动相中当甲醇比例较高时(如90%甲醇),4 种三嗪类农药出峰快;水体积相对较少时,如80∶20,则4 种三嗪类农药保留时间偏长;当甲醇-水(86∶14,V/V)时,4 种三嗪类农药保留时间适宜,且分离效果好,如图8所示。因此,本实验选择甲醇-水(86∶14,V/V)作为流动相。
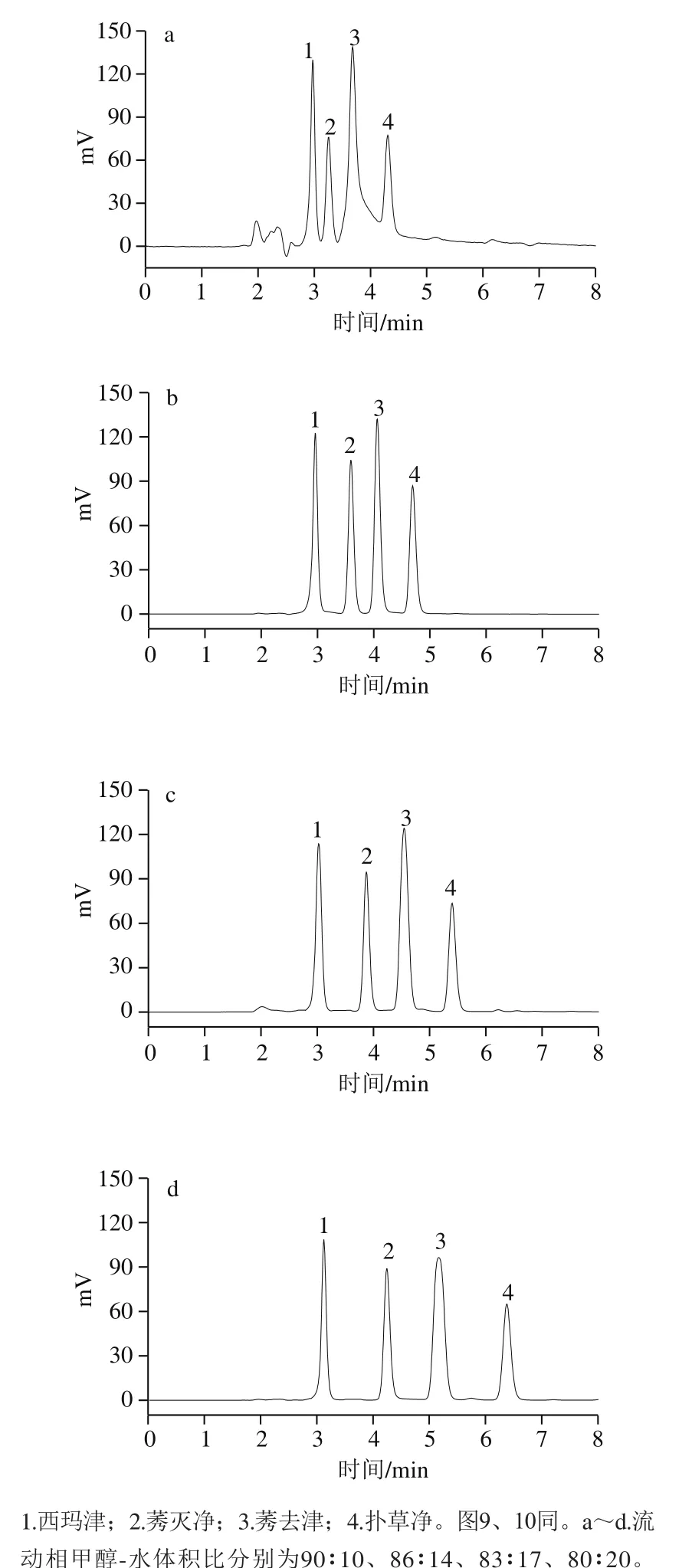
图8 4 种三嗪类农药分离过程中流动相比例的选择Fig. 8 Selection of mobile phase composition for separation of four triazine herbicides
2.6 莠去津-MISPE-HPLC检测食品中三嗪类农药残留
2.6.1 线性关系与检出限
分别配制4 种三嗪类农药质量浓度为0.5~50 μg/mL的一系列标准溶液,在1.3.6节色谱条件下对配制的溶液进行测定。以待测物质量为横坐标,峰面积为纵坐标作图,得到线性关系曲线,结果如表3所示。

表3 4 种三嗪类农药线性关系及检出限Table 3 Linear relationship and detection limits for four triazine herbicides
2.6.2 莠去津固相萃取柱对样品的净化和吸附能力
为考察莠去津固相萃取柱对样品中4 种三嗪类农药的净化吸附能力,实验选择多种样品进行测定,按照1.3.1节对样品进行提取,样品提取液和样品加标提取液用分子印迹固相萃取柱进行处理,然后用HPLC仪检测。由图9和图10a、b可知,在样品中未检出目标物。

图9 小麦样品提取液(a)、过MISPE洗脱液(b)和样品加标提取液过MISPE洗脱液(c)色谱图Fig. 9 Chromatograms of wheat extract (a), eluate from MISPE (b)and spiked eluate from MISPE (c)
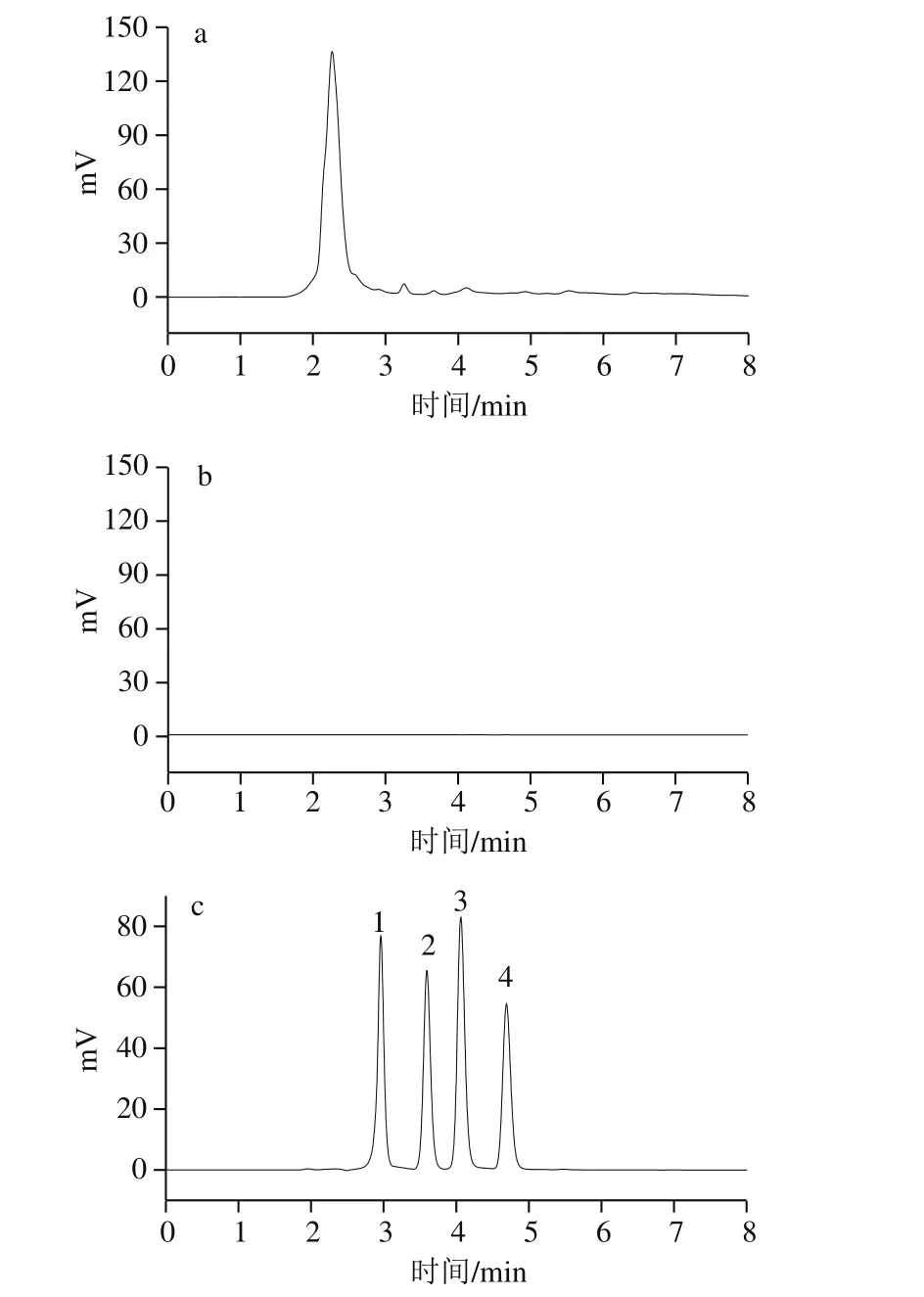
图10 苹果样品提取液(a)、过MISPE洗脱液(b)和样品加标提取液过MISPE洗脱液(c)色谱图Fig. 10 Chromatograms of apple extract (a), eluate from MISPE (b)and spiked eluate from MISPE (c)
同时,从图9、10还可以看出,该固相萃取柱可以从复杂基质中特异性分离富集目标物,对样品净化效果好(比较图9和图10a、b),该萃取柱只对目标物质及其结构类似物有选择性,而对杂质不具有吸附能力(比较图9和图10a、c)。
2.6.3 样品回收率和精密度实验
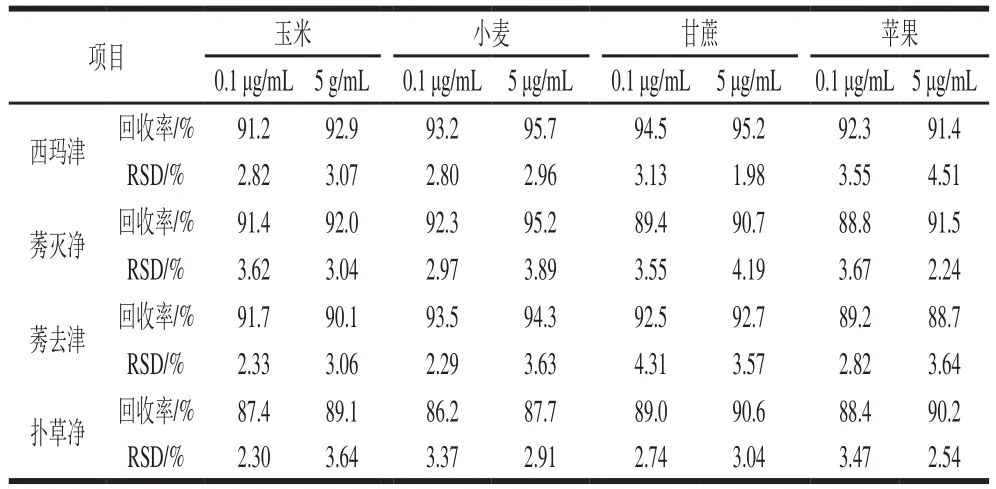
表4 回收率和精密度实验结果(n=5)Table 4 Recovery and precision of the method (n= 5)
为考察莠去津-MISPE-HPLC方法准确性和重复性,本实验选取玉米、小麦、甘蔗和苹果样品,分别在0.1 μg/mL和5 μg/mL加标量条件下进行回收率实验,结果见表4。样品中4 种三嗪类农药的加标平均回收率在86.2%~95.7%之间,相对标准偏差(relative standard deviation,RSD)为1.98%~4.51%(n=5),说明本方法回收率高、精密度好,基本满足农药残留分析要求。
3 结 论
采用沉淀聚合法合成了莠去津MIPs微球,并优化了聚合工艺。当莠去津和MAA物质的量比1∶4、致孔剂乙腈用量50 mL、聚合温度60 ℃时,制备的莠去津MIPMs吸附效果最好。静态吸附结果表明,所制得的MIPMs对模板物质具有较强的吸附能力;由Scatchard分析可知,莠去津和功能单体MAA形成了两类结合位点。竞争性吸附实验结果表明MIPMs不仅对模板物质莠去津有很好的吸附能力,对其他3 种结构类似物西玛津、莠灭净和扑草净均有吸附,吸附能力依次为莠去津>西玛津>莠灭净>扑草净。将莠去津MIPMs作为固相萃取填料制备了MISPE柱,建立了莠去津-MISPEHPLC检测食品中4 种三嗪类农药残留的方法。结果表明,莠去津MISPE柱对三嗪类农药残留有很好的吸附富集能力和样品净化效果,样品加标平均回收率为86.2%~95.7%, RSD不高于4.51%(n=5),检出限为0.5~5 ng/mL,基本满足农药残留分析要求。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
化工设计通讯(2020年4期)2020-01-15
酿酒科技(2019年10期)2019-11-12
小学生优秀作文(高年级)(2018年4期)2018-09-11
中国塑料(2015年4期)2015-10-14
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
