
可变性红斑角化症一例
2019-10-22 05:17郑晓丽梁官钊徐秀莲尹跃平刘维达
中国麻风皮肤病杂志 2019年10期
郑晓丽 梁官钊 徐秀莲 尹跃平 刘维达
作者单位:中国医学科学院&北京协和医学院皮肤病研究所,南京,210042
临床资料患者,男,10岁。因“周身干燥、起红斑10年”就诊。父母诉患者自出生起,躯干、四肢、臀部即出现大片淡红色斑片,伴边缘性脱屑,渐波及全身;部分皮损形状几天内可变化,扩大、消退同时存在;部分皮损持久存在,变化不明显;自觉病情冬季较夏季重,轻度瘙痒。父母否认家族类似患者,否认近亲婚配。平素身体良好,否认系统疾病。皮肤科查体:胸腹部、背部、臀部及四肢皮肤干燥,弥漫性淡红色斑或淡褐色斑,形态奇特如地图,边缘较清,皮损轻度脱屑;双下肢、腹部及胸部皮损左右对称,皮损间隙可见正常皮肤(图1);双手、足掌及面颈部未见明显异常。背部皮损组织病理示:表皮角化过度和角化不全,棘层轻度肥厚,轻度乳头瘤样增生,真皮乳头延长,真皮轻度水肿和少量炎细胞浸润(图2)。
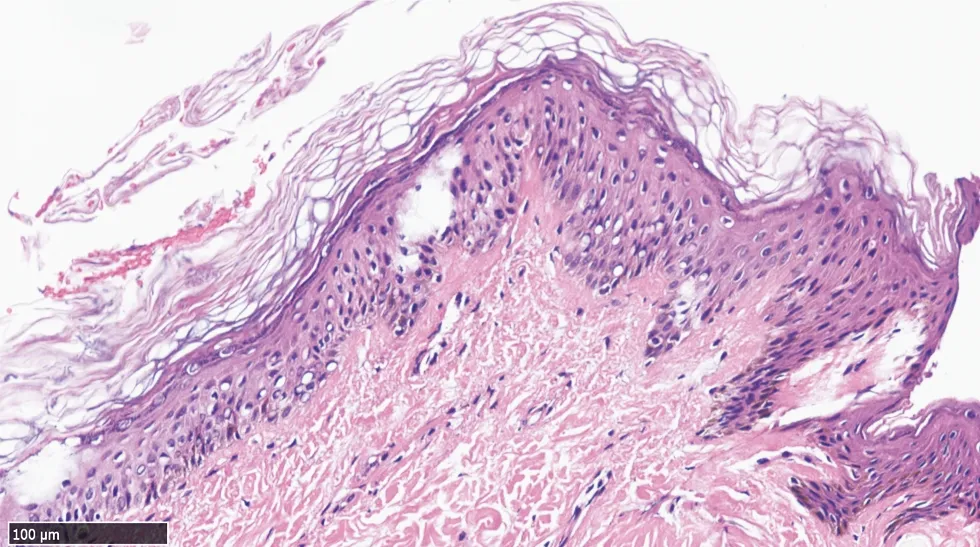
图2表皮角化过度和角化不全,棘层轻度肥厚,轻度乳头瘤样增生,真皮乳头延长,真皮轻度水肿和少量炎细胞浸润(HE,×100)
诊断:可变性红斑角化症。
治疗:予尿囊素乳膏洗澡后外用,维A酸软膏每晚一次外用。治疗后皮损有所好转,目前失访。
讨论1991年,Macfarlane等认为可变性红斑角化症(erythrokeratodermia variabilis, EKV)和进行性对称性红斑角化症 (progressive symmetric erythrokeratodermia, PSEK)具有相同遗传背景,仅临床表现不同。2009年,van Steensel等提出可变性和进行性红斑角化症(erythrokeratodermia variabilis et progressiva, EKVP),包括EKV和PSEK。OMIM网站将EKVP分5型:EKVP1,由GJB3(1p34.3)突变引起,有常染色体显性遗传(AD)和常染色体隐性遗传(AR)两种报道;EKVP2,由GJB4(1p34.3)突变引起,常染色体显性遗传;EKVP3,由GJA1 (6q22.31)突变引起,常染色体显性遗传[1];EKVP4,由KDSR(18q21.33)突变引起,常染色体隐性遗传[2];EKVP5,由KRT83(12q13.13)突变引起,常染色体隐性遗传[3]。
EKV在1925年由Mendes da Costa首次报道,故又称Mendes da Costa病。1978年赵天恩等国内首报,1984年van der Schroeff发现EKV与Rh(1p36-34)区域基因突变相关,1997年Richard等将致病基因更加精确的定位在1p34-p35,1998年Richard等发现GJB3突变,2000年Macari等报道GJB4突变,2015年Boyden发现GJA1突变。EKV发病年龄一般为出生时或出生后不久至3岁内发病,儿童期加重,偶见青春期发病,随着年龄增长有某些改善。皮疹特点表现为边界清楚的红斑性和角化过度性斑片,形态奇特。皮损可发生于任何部位,但多见于四肢伸侧、臀部、腋下、腹股沟和面部,50%并发掌跖角化症,并且有多汗,呈红斑脱屑状。经典EKV有两种类型皮肤损害:持久性角化过度性斑块和迁移性红斑。致病基因为编码间隙连接蛋白的基因,包括GJB3基因(编码连接蛋白31)和GJB4基因(编码连接蛋白30.3)。
本病需与泛发性体癣、Netherton综合征、非大疱性先天性鱼鳞病样红皮症相鉴别。泛发性体癣皮损初为圆形或椭圆形红色鳞屑性斑片或斑块,离心性扩散,可伴有瘙痒,随后皮损中心消退,而边缘仍呈活动性、隆起并继续向外扩展,形成环形离心性斑块,全身泛发时多个斑块可融合。Netherton综合征是一种罕见常染色体隐性遗传病,由SPINK5基因突变所致,表现为先天性伴有细小鳞屑的泛发性红皮病、迂曲性线状鱼鳞病、竹节样头发,常有生后最初几年生长迟缓。非大疱性先天性鱼鳞病样红皮症出生时即有皮肤发红,全身鳞屑如铠甲状,鳞屑脱落后,留下湿润面,四肢屈侧和皱襞部受累较重,可见灰棕色疣状鳞屑,随年龄增长,症状可逐渐减轻。
笔者对国内既往报道的可变性红斑角化症病例进行了总结和统计,以“可变性红斑角化症”、“可变性红斑角皮症”、“可变性红斑角化病”为检索词,检索自1994-2018年文献共31篇,病例数57例,准确记录发病年龄的病例数为35例,统计发现发病年龄<1岁者占62.8%(22/35),1~3岁占8.6%(3/35),3岁以后占28.6%(10/35),其中最小发病年龄为出生1个月内,最大发病年龄为40岁。男性占56.6%(30/53),女性占43.4%(23/53),余4例未报道患者性别。国内大多数病例报道未行基因检测,仅4篇文献进行了基因检测,其中两篇未检测出GJB3和GJB4突变,另2篇测出的基因突变有GJB3 L135V、GJB3 c.324G
猜你喜欢
中国美容医学(2022年2期)2022-03-17
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
百科探秘·航空航天(2018年11期)2018-11-29
家庭医学·下半月(2018年8期)2018-10-17
家庭医药(2018年2期)2018-02-09
军事文摘(2017年22期)2017-11-01
东方教育(2017年14期)2017-09-25
家庭医药(2016年4期)2016-05-04
课程教育研究·学法教法研究(2016年1期)2016-03-17
