
AMG 510只是攻克RAS的一小步(一)
2019-10-18 01:11谢雨礼
国际人才交流 2019年10期
文/谢雨礼
(谢雨礼,毕业于南开大学化学系,获
得中国科学院上海药物研究所博士学位,在哥伦比亚大学从事博士后研究工作。2017年4月,创办苏州偶领生物医药有限公司,致力于创新化药的研究和开发)
回顾2019年的行业热点,小分子药物的热度在免疫疗法的喧嚣过后似乎有所回升,其中最大的亮点就是安进公司的KRAS G12C小分子抑制剂AMG510。继ASCO后,9月中旬,安进在巴塞罗那的世界肺癌大会(WCLC)上更新了AMG510的一期临床数据:携带KRAS G12C突变的非小细胞肺癌病人中,960mg高剂量组ORR达到54%,疾病控制率100%,未见明显毒副反应。考虑到这些病人都是经过重度治疗的,这个数据算是比较亮丽的。其实,业界之所以感到兴奋,可能并不是因为这个数据有多好,而是RAS这个被认为不可成药的“致癌基因之王”,经过30年努力,其直接抑制剂第一次接近成功。
RAS是第一个被发现的人类肿瘤基因(oncogene)
为什么RAS在肿瘤中这么重要?这可能还得从肿瘤生物学的起源说起。19世纪末,巴斯德等巨匠的研究让现代医学在抗感染领域获得巨大成功。1876年,一个俄国人成功地将肿瘤组织从一只狗移植到另外一只狗。1908年,两个丹麦科学家发现,母鸡白血病细胞中的提取物可以让其他鸟类感染白血病。这些观察和研究让人们相信肿瘤也是病毒所致的传染病。1910年,洛克菲勒研究所的Peyton Rous从一只来自纽约长岛的母鸡中发现了第一个肿瘤病毒Rous Sarcoma Virus(RSV,劳氏肉瘤病毒)。此后,一系列导致动物肿瘤的RNA和DNA病毒相继被发现。致病机理也得到了合理的解释:这些病毒(RNA病毒通过逆转录酶)可以将肿瘤基因整合到被感染的宿主细胞中,诱导恶变并维持肿瘤细胞的无限分裂。发现RSV的Peyton Rous和发现RNA病毒逆转录酶的Howard Temin分别获得1966年和1975年的诺贝尔生理学或医学奖。至此,人们深信肿瘤是一种病毒导致的疾病。
直到1974年,UCSF的J. Michael Bishop和他的博士后Harold Varmus通过DNA探针意外发现未被感染的正常细胞中也存在此前RSV病毒中发现的肿瘤基因SRC。原来,肿瘤基因早就存在于宿主的基因组当中,远古时期,病毒从宿主细胞中获得这些基因片段并加以改造,也就是说,病毒所携带的肿瘤基因其实来自我们自身。这些发现打开了以基因变异为基础的现代肿瘤生物学的大门。Bishop和Varmus也因此在1989年获得了该领域的第三个诺贝尔奖。
Harvey和Kirsten等人在20世纪60年代分别发现了类似于RSV的逆转录病毒携带的老鼠肿瘤基因Hras和Kras。1982年,Weinberg等实验室在人类膀胱癌细胞T24/EJ中也发现了HRAS,使得RAS成为第一个被发现的人类肿瘤基因。50多年过去后,RAS基因被发现是肿瘤中最常见的突变基因之一,30%的肿瘤携带RAS变异,如果算上RAS调控因子和信号通路上下游的变异,几乎覆盖所有肿瘤,RAS变异每年造成100万以上的病人死亡,无愧是“肿瘤基因之王”。这也令人非常好奇,RAS到底具有什么神奇功能,令肿瘤细胞如此依赖。
RAS与肿瘤抑制基因pRb和p53一起成为肿瘤领域难以攻克的高地
谈到RAS的致癌功能,又要提起肿瘤发生机制的主要假说SMT(Somatic Mutation Theory)。该理论认为肿瘤来源于正常细胞,基因突变的驱动下,获得生存优势,不断进行适者生存的进化,最后演变成具有基因组不稳定、无限增殖和抗凋亡等十大标志的肿瘤细胞。这十大标志或者说能力都服务于肿瘤细胞的唯一目标:无限复制而获得永生。细胞的复制开始于休止期(G0),接收生长因子信号的刺激后,启动分为四个阶段(G1、S、G2和M)的细胞分裂周期。在这个周期中,母细胞一分为二,复制基因并均等地分配给两个子细胞。肿瘤基因突变或者异常可以驱动生长因子信号失控,造成细胞不受控制的生长和分裂。RAS处于各种信号通路的中枢位置,下游控制的通路包括Raf、PI3K和RalGDS等。RAS在分子水平上可以调控蛋白质的合成,基因转录等过程;细胞水平上影响细胞形状、生长、分化、凋亡、转移等等。生长因子受体EGFR、FGFR,以及转录因子Myc的变异也能驱动肿瘤的形成,RAS既可以在这些肿瘤基因的下游传递放大信号,也可以反馈增强它们的致癌功能。
如果肿瘤基因突变就轻易导致肿瘤,物种进化就不会保留它们。事实上,肿瘤的发生还需要对抗肿瘤抑制基因的作用。正常细胞的生长、分裂和增殖是由复杂信号网络所严格控制的。比如细胞周期中,遗传物质复制出错就可能被中止分裂。细胞周期中有一个重要节点叫作限制点(R point),处于G1期的中后段,细胞分裂一旦越过R点,就无法停止,即使没有生长因子的存在,细胞分裂也会进行到底。控制这个R点的关键基因叫作pRb。在CDK/Cyclin的调控下,磷酸化的pRb通过控制转录因子E2F决定细胞周期是否可以越过R点。pRb是限制细胞不正常分裂的最重要的检查点,自然也是至关重要的肿瘤抑制基因。与RAS一样,几乎所有肿瘤中,pRb的正常功能被抑制或者被破坏。另一个与pRb同等重要的肿瘤抑制基因是监控细胞健康状态的转录因子p53。正常细胞一旦受到破坏,p53会中止细胞的生长和分裂,启动修复机器,如果无法修复则启动细胞凋亡程序。生长因子失控或者是pRb功能缺失,也会导致p53介导的凋亡。大量突变和功能异常的肿瘤细胞如果希望永生,必须摆脱p53的控制,因此p53的功能丧失突变出现在三分之一的肿瘤之中,加上其相关通路的异常也是几乎覆盖100%的肿瘤。
这样,RAS过度激活以及pRb和p53功能丧失成为导致肿瘤的三座大山,它们是目前发现的肿瘤中突变最多的三个基因。这也说明它们处于肿瘤细胞信号网络的最关键节点上。广义上说,绝大多数靶向药物的靶点都处于这三个基因调节的信号通路中。俗话说擒贼先擒王,十分遗憾目前还没有直接靶向这三个靶点的药物出现。从生物学的角度上看,pRb和p53是肿瘤抑制基因,功能异常经常体现在蛋白缺失或者功能被抑制,难以用药物进行干预。近年来,也有人尝试用小分子纠正变异p53的构象,恢复其野生型的功能。相关药物APR-246和COTI-2已进入临床研究,但进展缓慢,有效性仍未得到验证。乍一看,RAS相对于pRb和p53,作为异常激活的肿瘤基因,是更加理想的药物靶点。事与愿违,经过多年的探索,RAS被认为是不可成药靶点,攻克RAS成为制药界的“哥德巴赫猜想”。
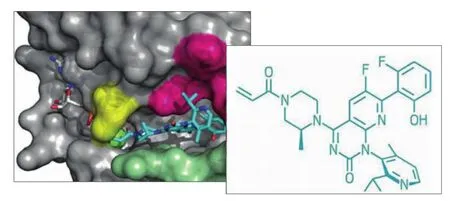
AMG-510与KRAS G12C复合物结构
RAS蛋白的结构、工作原理和调控机制
为什么RAS长期以来被认为不可成药?这要从其结构,工作原理和调控机制说起。RAS主要有三个亚型HRAS、KRAS和NRAS,其中,KRAS通过RNA剪接又表达两个不同的蛋白KRAS4A和KRAS4B。RAS蛋白是一种21kDa大小,位于细胞膜上的,拥有GTPase酶活性的鸟嘌呤核苷结合蛋白。RAS是GDP/GTP循环控制的二进制分子开关:GDP结合时处于“关”的状态,而GTP结合则代表“开”的状态。没有刺激信号时,这种关开状态的转换非常缓慢。接收到信号后,鸟苷酸交换因子GEFs被招募到细胞膜上与RAS结合并释放GDP。由于细胞中GTP的浓度远高于GDP,RAS蛋白迅速结合GTP,招募其下游信号通路的效应蛋白进入“开”的状态。在GAPs(GTPase Activating Protein)的作用下,RAS的GTPase的活性被提高上千倍,其结合的GTP被水解成GDP,RAS重新进入GDP结合的“关”的状态,从而完成一个完整的循环。
RAS分子开关的结构基础也得到了很好的阐释。RAS蛋白1—166个氨基酸组成G-domain,这个结构域在各亚型中高度保守,余下的23—24个氨基酸组成高度可变区(HVR)。D-domain带有RAS的核苷酸结合口袋,包括P-loop(残基10—17)、Switch I(残基30-38)和Switch II(残基60—67)等区域。从其名字可以判断,Switch I和II是RAS功能切换的关键。GTP结合的活性状态时,GTP分子中Gamma位磷酸分别与Switch I的T35和Switch II的G60形成氢键,像弹簧一样将它们拉在一起。一旦GTP被水解,Gamma位磷酸被水解掉,Switch I和II被分开,回到GDP结合的非活性构象。
从上述描述可以看出,GTP水解是淬灭RAS活性的关键步骤。不难想象,如果基因突变抑制了GTP水解,RAS蛋白将被锁在激活状态。正因为如此,RAS最常见的突变就发生在G12、G13和Q61三个位置。前面提到RAS高效水解GTP需要GAP蛋白参与,GAP与RAS结合后,将一个精氨酸(arginine)残基伸到催化位点,协助水解,这个残基被称作精氨酸手指(arginine finger)。结构生物学研究表明,除了脯氨酸,G12或G13替换成任何氨基酸,立体位阻将阻碍精氨酸手指进入催化位点。Q61则是直接参与水解过程,协助定位一个水分子,稳定水解反应的过渡态。其他突变还有与GDP结合相关的氨基酸如A146。比较有意思的是,虽然各个亚型的D-domain和功能高度类似,但肿瘤中发现的RAS突变主要集中在KRAS(85%),NRAS(12%)次之,HRAS(3%)最少。最近的研究也许提供了部分解释,他们发现KRAS4B而不是其他亚型通过HVR区域与calmodulin结合,从而抑制非经典Wnt通路,有利于保持肿瘤细胞的干性状态。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
中国现代医生(2022年19期)2022-08-25
中国典型病例大全(2022年9期)2022-04-19
保健与生活(2021年24期)2021-12-12
中学化学(2019年3期)2019-07-08
健康大视野(2018年6期)2018-07-17
中学化学(2016年2期)2016-05-31
课程教育研究·下(2016年2期)2016-03-25
销售与管理(2006年9期)2006-09-17
