
无硫高膨胀体积膨胀石墨的制备
2019-10-14 08:53田啊林黄雪莉胡子昭王雪枫
无机盐工业 2019年10期
田啊林,黄雪莉,胡子昭,王雪枫
(新疆煤炭洁净转化与化工过程重点实验室,新疆大学化学化工学院,新疆乌鲁木齐830046)
膨胀石墨(EG)因具有比表面积大、化学稳定性好、高导电导热、耐高低温、耐腐蚀、阻燃、可回弹等诸多优良性能,在密封、环保、防火阻燃、生物医学、军事等多个领域中得到应用,成为重要的功能石墨材料之一[1-3]。由于膨胀石墨中残余的硫易造成金属腐蚀、催化剂失活等使得其在密封材料及催化剂载体中的应用受到影响[4-6],所以低硫或无硫的膨胀石墨成为研究的热点。其主要采用不含硫的HClO4、KMnO4、K2Cr2O7、H2O2等为助氧化剂,以汽化温度较低的冰乙酸等为插层剂,在酸性条件下对粒径为300 μm左右的鳞片石墨(FG)进行氧化,所制得的膨胀石墨容积为 220~510 mL/g,膨胀温度在800~1000℃[7-10]。这些方法具有能耗高、产物膨胀体积小、环境污染大等缺点,使EG的特性难以充分发挥。大鳞片石墨资源日益减少,来源受限,以小鳞片石墨为原料制备的无硫、高膨胀体积且具有更低膨胀温度的膨胀石墨成为人们追求的目标[11-14]。
本文以高氯酸-磷酸-乙酸酐混酸为反应体系,采用混合固体氧化剂为助氧化剂,以粒径为180 μm的鳞片石墨为原料,在500℃马弗炉上膨化制备出膨胀体积高达665 mL/g的无硫膨胀石墨。
1 实验部分
1.1 原料试剂及仪器
原料及试剂:粒径为180 μm的天然鳞片石墨(纯度为98%)、乙酸酐(质量分数为98.5%)、磷酸(质量分数为85%)、高氯酸(质量分数为70%)、过氧化氢(质量分数为30%)、高锰酸钾、重铬酸钾,以上均为分析纯。
仪器:D8 ADVANCE型X射线衍射仪,CuKα辐射,扫描范围 2θ为 10~80°;VERTEX70 型红外光谱仪;STA 449C型热分析仪,氩气气氛,升温速率为10℃/min;HH-S4型数显恒温水浴锅;DHG-9015A型电热鼓风干燥箱;KSL-1200X型马弗炉;DL-1型实验电炉;GL224-1SCN型电子分析天平。
1.2 实验步骤
1)混酸的配制。按照弱酸到强酸的顺序,依次向置于冰水浴中的棕色试剂瓶内加入13 mL乙酸酐、12 mL磷酸和17 mL高氯酸,在加入过程中不断搅拌至均匀。
2)可膨胀石墨的制备。取1 g FG于烧杯中,加入4 mL混酸,之后依次加入0.1 g高锰酸钾和0.1 g重铬酸钾,搅拌均匀后置于恒温水浴锅内,60 min后取出。向烧杯内加入适量的过氧化氢,反应一定时间后至溶液无色时停止加入。经离心洗涤后,于40℃干燥箱内干燥2 h即得可膨胀石墨(GICs)。
3)膨胀石墨的制备。取0.1gGICs置于已在电炉上烧至恒温的坩埚内,焙烧至体积不再发生变化为止,即可制得EG。冷却至常温,对膨胀体积进行测定。
4)膨胀体积的测定。采用标准量筒法按公式(1)测膨胀体积(EV)[15-16],每个样测量至少3次取平均值。

式中:V为膨胀后试样的体积,mL;m为膨胀后试样的质量,g。
2 结果与讨论
2.1 膨胀石墨的制备条件对膨胀体积的影响
2.1.1 混合固体助氧化剂配比对膨胀石墨膨胀体积的影响
混合固体助氧化剂配比对膨胀石墨膨胀体积的影响如表1所示。由表1可知,随着高锰酸钾与重铬酸钾质量比的增加,EG的膨胀体积先增大后减小,在m(KMnO4)∶m(K2Cr2O7)=1∶1 时达到最大,为 594 mL/g。KMnO4与K2Cr2O7均是强氧化剂,在酸性溶液中的电极电位可通过式(2)、(3)进行计算:
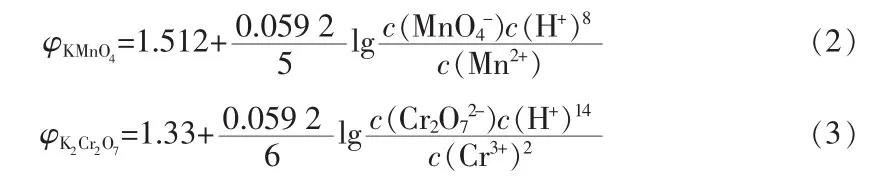
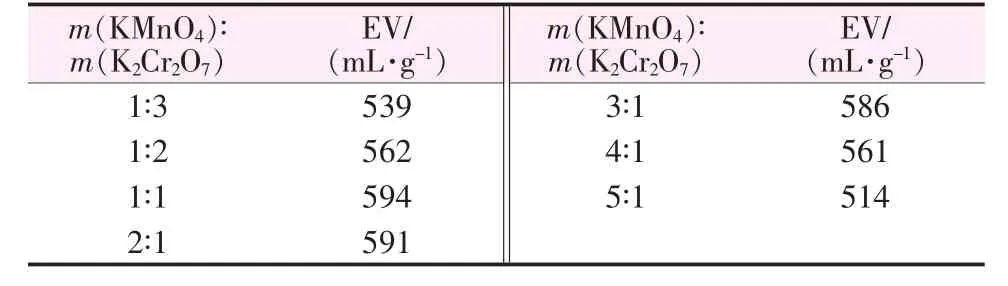
表1 混合固体助氧化剂配比对膨胀石墨膨胀体积的影响
本文通过加入乙酸酐吸收溶液中多余的水分,大大提高了溶液中H+的浓度,使整个反应体系氧化性提高,且经计算得 φKMnO4=1.64,φK2Cr2O7=1.52,KMnO4的电极电位高于K2Cr2O7,在氧化作用的增强方面占主导作用。因此,当m(KMnO4)∶m(K2Cr2O7)处于 1∶3~1∶1时,膨胀体积随着高锰酸钾用量的增加而增加;之后随着高锰酸钾用量的继续增加,氧化性随之增强,过氧化现象[5]逐渐突出,造成EG的膨胀体积减小。
2.1.2 混酸用量对膨胀石墨膨胀体积的影响
混酸用量对膨胀石墨膨胀体积的影响见表2。如表2所示,EG的膨胀体积随混酸用量的增加呈现先升高后降低的趋势。当混酸用量为4 mL时,膨胀体积达到最大值,为562 mL/g。
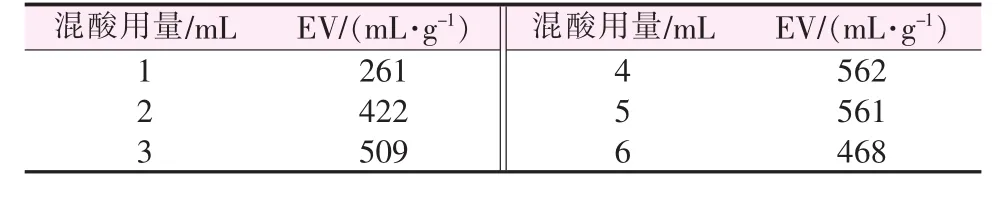
表2 混酸用量对膨胀石墨膨胀体积的影响
由实验现象可知,当混酸用量为2 mL(经换算为3.1 g)时,FG能够刚好浸润,与混合氧化剂进行氧化反应,膨胀体积能够达到422 mL/g,远少于文献[8,17]中同等膨胀体积下的酸用量。随着酸用量的增加,HClO4、H3PO4等氧化剂与FG接触越充分,石墨层间距打开越充分,进入了更多的插层剂,使膨胀效果更好。但当混酸用量超过5 mL/g时,此时体系中的高锰酸根和重铬酸根的浓度逐渐降低,整个体系氧化作用逐渐减弱,影响插层作用,故而膨胀体积随之减小。
2.1.3 反应温度对膨胀石墨膨胀体积的影响
反应温度对膨胀石墨膨胀体积的影响见表3。如表3所示,膨胀体积随反应温度的增加先增后减。由于低温时高氯酸氧化性较弱,而磷酸基本不显氧化性,随着温度的增加高氯酸和磷酸的氧化性逐渐增强,与此同时混合固体助氧化剂的溶解度也会随之增加,因此整个反应体系的氧化性及反应速率均得到增加,在50℃时膨胀石墨达到最大膨胀体积553mL/g。但因石墨的酸化氧化反应属于放热反应,进一步增加体系温度会抑制反应的正向进行,而且部分插层剂汽化,影响插层效果,导致膨胀体积变小。
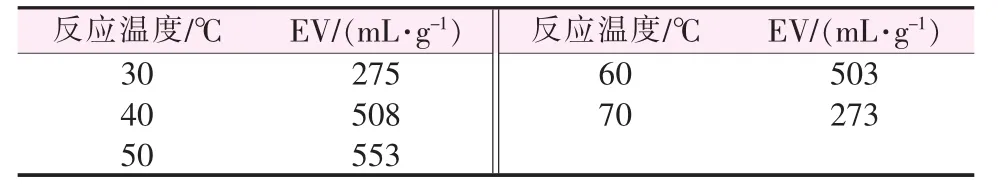
表3 反应温度对膨胀石墨膨胀体积的影响
2.1.4 反应时间对膨胀石墨膨胀体积的影响
反应时间对膨胀石墨膨胀体积的影响见表4。如表4所示,反应体系在反应到60 min时就达到了最大膨胀体积553 mL/g,混合固体助氧化剂的加入大大增加了反应速率,缩短了最佳反应时间,降低了时间成本。原料为同样粒径的FG,EG最大膨胀体积是文献[18]的两倍。但当反应时间超过70 min后,膨胀体积随时间的增加而降低,这是由于层间插层物与溶液中的插层物形成浓度差,随着时间的增加插层物向溶液中扩散的越多,层间插层物逐渐减少,高温焙烧时汽化产生的推动力减少,继而膨胀体积减小。

表4 反应时间对膨胀石墨膨胀体积的影响
2.1.5 混合固体助氧化剂用量对膨胀石墨膨胀体积的影响
混合固体助氧化剂用量对膨胀石墨膨胀体积的影响见表5。如表5所示,膨胀体积随着混合固体助氧化剂用量的增加先增大后减小,且变化趋势很明显。这主要是因为高锰酸钾和重铬酸钾本身氧化性较强,加之二者在强混酸溶液中的氧化性进一步被强化[8,18-19]。当混合固体助氧化剂用量小于 0.2 g时,膨胀石墨的膨胀体积随混合固体助氧化剂用量的增加而增加,但当混和固体助氧化剂用量超过0.23 g时,膨胀体积呈现锐减趋势,这是由于本体系的氧化强度较大,易造成鳞片石墨的过氧化[5],使可膨胀石墨粉末化严重,从而膨胀体积减小。

表5 混合固体助氧化剂用量对膨胀石墨膨胀体积的影响
2.1.6 干燥温度对膨胀石墨膨胀体积的影响
干燥温度对膨胀石墨膨胀体积的影响见表6。文献[20]提到水分有助于膨胀石墨低温孔隙结构的形成。如表6所示,当干燥温度低于40℃时,可膨胀石墨的含水量过多,焙烧时吸收大量热,降低实际膨化温度,减少膨化推动力,导致膨胀体积较小;随着干燥温度提高,剩余的石墨层间水对膨胀体积的增大起到了促进作用,在40℃时膨胀体积高达575 mL/g;但继续提高干燥温度,石墨层间水全部蒸发,没有额外的膨胀推动力,导致膨胀体积随膨胀温度的变化很小。
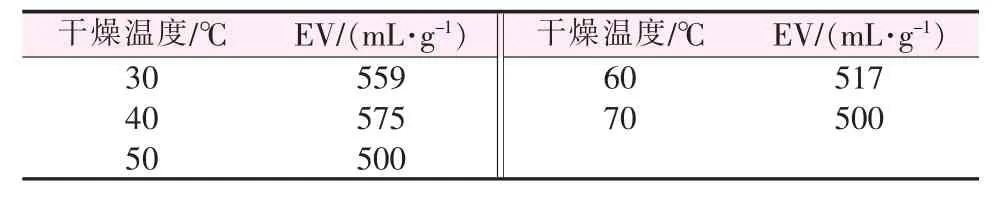
表6 干燥温度对膨胀石墨膨胀体积的影响
2.1.7 干燥时间对膨胀石墨膨胀体积的影响
干燥时间对膨胀石墨膨胀体积的影响见表7。如表7所示,干燥温度为40℃时,膨胀石墨的膨胀体积随干燥时间的增加先升高后降低,干燥时间为2 h时膨胀体积达到最大值585 mL/g,说明此时层间插层物汽化推动力最大。
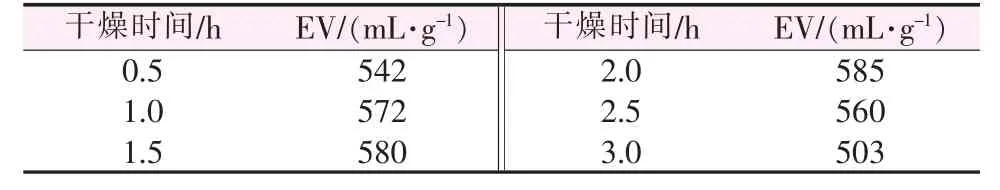
表7 干燥时间对膨胀石墨膨胀体积的影响
2.1.8 膨胀温度对膨胀石墨膨胀体积的影响
膨胀温度对膨胀石墨膨胀体积的影响见表8。从表8可知,该体系制备的GICs在200℃即可获得膨胀体积为79 mL/g无硫膨胀石墨,这是由于插层剂高氯酸、磷酸、乙酸酐的分解温度较低,通过FT-IR、XRD表征证明均已进入石墨层间[21-22]。随着膨胀温度的增加,石墨层间插层物汽化所产生推动力越大,膨胀体积也就不断增大[23]。当膨胀温度大于500℃后,膨胀体积变化不大,说明插层剂已经基本汽化完毕。
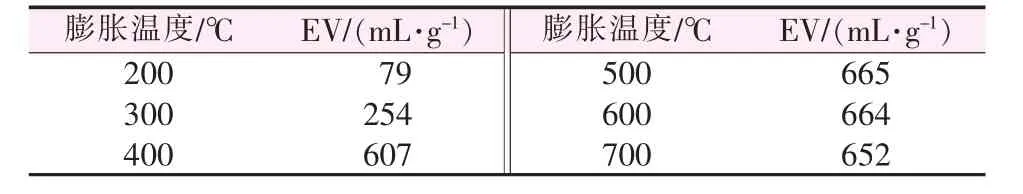
表8 膨胀温度对膨胀石墨膨胀体积的影响
2.2 FG、GICs及EG的SEM图
图1为 FG、GICs、EG 的 SEM 图。从图1a、1b可以看出,天然鳞片石墨表面平滑,石墨层紧密。FG经过氧化后,石墨表面变粗糙,从图1b和图1d对比发现石墨片层已被打开。再经电炉焙烧膨胀,形成如图1e的蠕虫状膨胀石墨,从图1f膨胀石墨的放大SEM图可以发现该法制备的膨胀石墨具有较大的缠绕空间、明显的V型裂开结构和网络型开放孔隙结构[24],说明插层反应充分,膨胀较完全。

图1 FG(a、b)、GICs(c、d)、EG(e、f)的 SEM 图
2.3 FG、GICs及 EG的XRD图
原料FG、最佳反应条件下制备的GICs及用电炉焙烧得到的EG的XRD谱图如图2所示。由图2可知,制得的GICs最强衍射峰的位置、强度和峰形与FG对比均有改变,其位置由FG的2θ=26.741°(004)左移到 2θ=25.111°处;峰型变宽,晶面间距由原来的d=0.33310 nm扩大到d=0.35434 nm;最强衍射峰强度变大。其次,从GICs的XRD图上发现3处新增的衍射峰位置分别在2θ为30.216、51.510、57.063°处,结合SEM图发现致密结构的石墨层被打开,说明插层剂已成功进入石墨层间。经膨胀后,发现EG在2θ=30.216°处的衍射峰消失,最强衍射峰位置又回到2θ=26.556°处,基本与FG重合,说明插层剂全部汽化,得到成分较纯的EG,且石墨内部的晶体结构未发生变化。
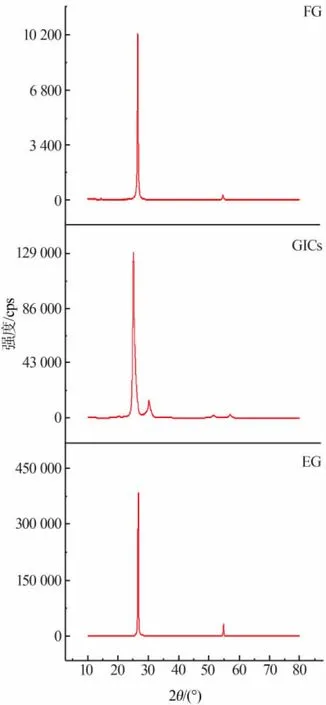
图2 FG、GICs、EG 的 XRD 图
2.4 FG、GICs、EG 的 FT-IR 图
图3 为 FG(a)、GICs(b)、EG(c)的红外光谱图。从图3b可知,FG经过该体系反应后,在3134.08 cm-1处出现了微弱的—OH伸缩振动宽吸收峰,说明GICs层内有少量的水存在;在1083.76 cm-1处出现的较强较宽的吸收峰,则是C—O键和—ClO4-官能团共同作用的效果;627.37 cm-1处对应的是Cl—O键;在 1501.22、866.79、537.83 cm-1处出现 C—O 伸缩振动峰;在1637.70 cm-1处出现—COOH的伸缩振动峰,证明高氯酸、乙酸酐已成功插层。经过高温焙烧后,从图3c可知,大部分新增振动峰消失或强度减弱,剩下1024.03cm-1处的C—O键与418.36cm-1处的C—C单键振动峰,说明瞬时高温致使插层物汽化作为膨胀推动力。

图3 FG(a)、GICs(b)和 EG(c)的 FT-IR 图
2.5 GICs的热重图
在氩气气氛中测得可膨胀石墨的热重图见图4。从图4可明显看出,在200℃附近出现了一个明显的质量损失平台,所对应的DSC曲线出现一个吸热峰,这是200℃时高氯酸和水分的汽化导致的5%的质量损失。200℃之后,质量损失率开始增大,说明大部分的插层物开始汽化或分解,400℃以后质量损失率基本为零。这一点与表8的结果基本吻合。
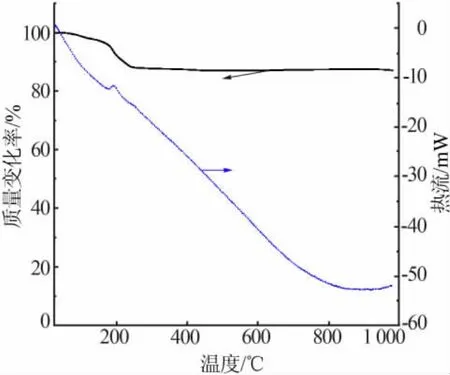
图4 GICs的热重图
3 结论
本文对混酸混合固体助氧化剂体系下制备的膨胀石墨进行了参数的筛选和优化,并研究了不同阶段的产物特征,获得如下结论:1)按m(FG)∶V(混酸)∶m(混合固体助氧化剂)=1(g)∶4(mL)∶0.2(g),其中混合固体助氧化剂中m(KMnO4)∶m(K2Cr2O7)=1∶1,反应温度为 50℃,反应时间为 60 min,干燥温度为40℃,干燥时间为2 h的条件合成的可膨胀石墨具有低初始膨胀温度,并且在500℃能够获得膨胀体积高达665 mL/g的无硫膨胀石墨。2)经SEM、XRD、FT-IR及TGA表征,表明FG经氧化插层后,石墨边缘层被打开,高氯酸根、磷酸根及乙酸酐插入了层间,导致晶体结构改变。经膨胀后,插层物汽化,晶体结构趋于完整,制得成分较纯、缠绕空间大、孔隙结构丰富的蠕虫状无硫高倍膨胀石墨,其在作为吸附材料、载体及制备石墨烯前驱体材料等方面具有较高的市场应用价值。
猜你喜欢
河南科技(2022年8期)2022-05-31
昆钢科技(2022年1期)2022-04-19
食品安全导刊(2021年20期)2021-08-30
纺织科学研究(2021年7期)2021-08-14
军事文摘(2020年20期)2020-11-16
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
安徽农业科学(2018年36期)2018-05-14
中学物理·初中(2017年12期)2018-03-07
科技创新导报(2017年16期)2017-08-23
