
拷贝数变异检测算法优化研究
2019-10-08 09:01林勇
软件 2019年3期
林勇

摘 要: 拷贝数变异与多种复杂疾病密切相关,具有重要的研究意义。本文利用基于测序数据的拷贝数变异检测过程中丢弃的不匹配读数据,采用裂读法和单端匹配法对已有检测算法的结果进行过滤优化。模拟和实验数据检测结果表明,本文方法优化后能得到了更高的检测性能。
关键词: 拷贝数变异检测;算法优化;裂读法; 配对末端读数
【Abstract】: Copy number variation is closely related to many complex diseases and has important research significance. In this paper, the mismatched read data discarded in the process of copy number variation detection based on sequencing data were used to filter and optimize the results of existing detection algorithms by split reading method and one end matching method. The detection results of simulation and experimental data showed that the proposed method can achieve higher detection performance after optimization.
【Key words】: Copy number variant detection; Algorithm optimization; Split read; Paired-end read
0 引言
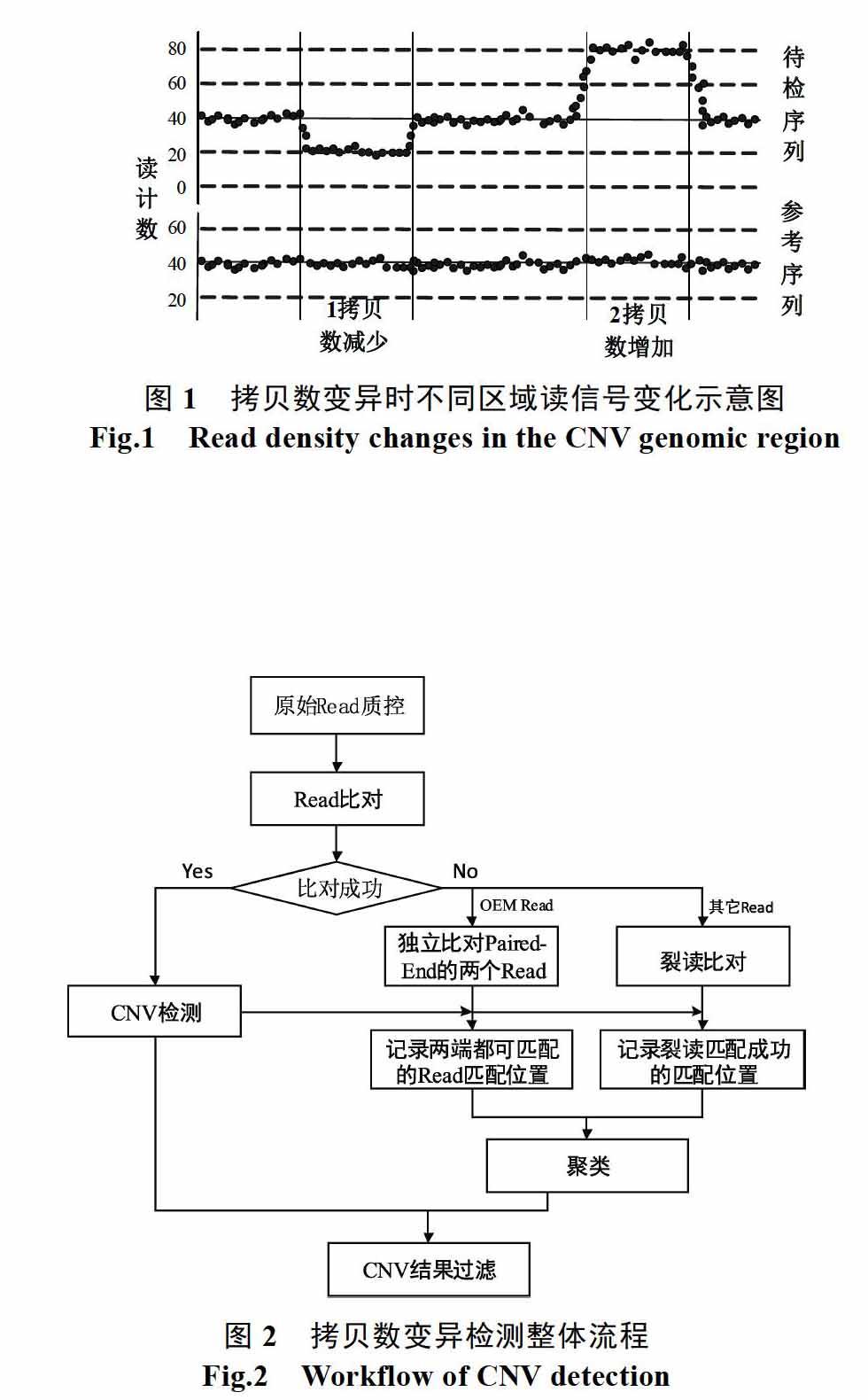
拷贝数变异(Copy Number Variation, CNV)是大小超过1 kb的亚显微突变,表现为DNA片段缺失或重复,重复又分为散落重复和串联重复。拷贝数变异广泛地存在于人类基因组上[1],雖然拷贝数变异的频率较低,但累积的碱基数量却大大超过单核苷酸多态[2-4]。许多研究证明了拷贝数变异与包括乳腺癌、孤独症、肥胖和骨质疏松症等多种疾病相关[5-8]。随着下一代测序技术(Next-Generation Sequencing, NGS)的出现和发展,拷贝数变异检测也出现了全新的方法,下一代测序技术能够检测DNA序列上碱基量级的序列信息,通过对测序数据进行分析和检测,拷贝数变异长度、位置等信息就的可能检测获得。现有的基于测序数据的拷贝数变异检测方法主要是基于读深度法(Read-Depth Method),它的基本原理是基于拷贝数的变化将引起测序数据匹配区域的读数据累计量会出现显著地增加或减少。如图1所示。目前,研究人员已经开发了许多基于度深度法的检测工具,比如:CNV-seq[9],ReadDepth[10],CONTRA[11]和CNVnator[12]。
目前常规方法检测过程中,首先将读数据与 参考序列进行比对,由于读数据存在少量的测序错误以及序列上的SNP问题,比对时允许少量碱基的错配,当大于错配阈值时,读数据将被丢弃。实际上这些被丢弃的读数据并不都是“垃圾数据”,本文对读数据匹配不成功的成因进行过分析,加以分类,记录裂读匹配(Split Read Match,SRM)和单端匹配(One End Match,OEM)的数据,对匹配位置进行聚类后对拷贝数检测的初步结果进行 筛选,从而得到更精确的检测结果,从而提高检测性能。
1 基于OEM和SRM的拷贝数变异检测优化
本文检测拷贝数变异的整体流程如图2所示,该方法仅针对末端配对(Paired-End)的Read数据进行CNV检测,目前通用的测序仪产生的Read以Paired-End数据为主,因此本文算法适用范围较广。首先对原始Read数据进行质控,过滤掉Q值低于20的Read,然后将质控结束后得到的read数据与参考序列进行比对,本文采用的比对工具为BWA[13],比对得到的数据通过samtools转换成SAM文件,便于分析比对结果。比对得到的结果分为两部分:
(1)匹配成功的Read数据用于初步的拷贝数变异检测,本文采用的拷贝数检测工具为CNVnator[14],CNVnator通过检测序列的深度分布情况,结合GC校正、均值漂移法、多带宽分割等方法进行拷贝数变异的检测,是目前较常用的一种基于读深度法检测拷贝数变异的工具。在检测过程中,我们将信号强度阈值降低,这有利于检测出更多可能的CNV。检测获得的结果作为候选结果用于后期筛选;
(2)匹配成功的Read数据,又分为两种,一种是OEM Read,是指Paired-End Read的两个单端皆能与参考序列匹配但匹配位置距离与Paired-End的插入距离(insert length)差异很大,因此在序列比对的时候被丢弃的Paired-End Read;另一种是非OEM Read,对于这类Read我们进行裂读处理,然后将裂读后的数据进行再比对,获得裂读匹配位置。
接着我们对上述获得的两种匹配位置进行聚类,最后使用聚类结果完成对候选CNV的筛选得到最后的CNV检测结果。下面我们对OEM、SRM和聚类分析的工作原理和技术细节进行详细的描述。
1.1 OEM检测
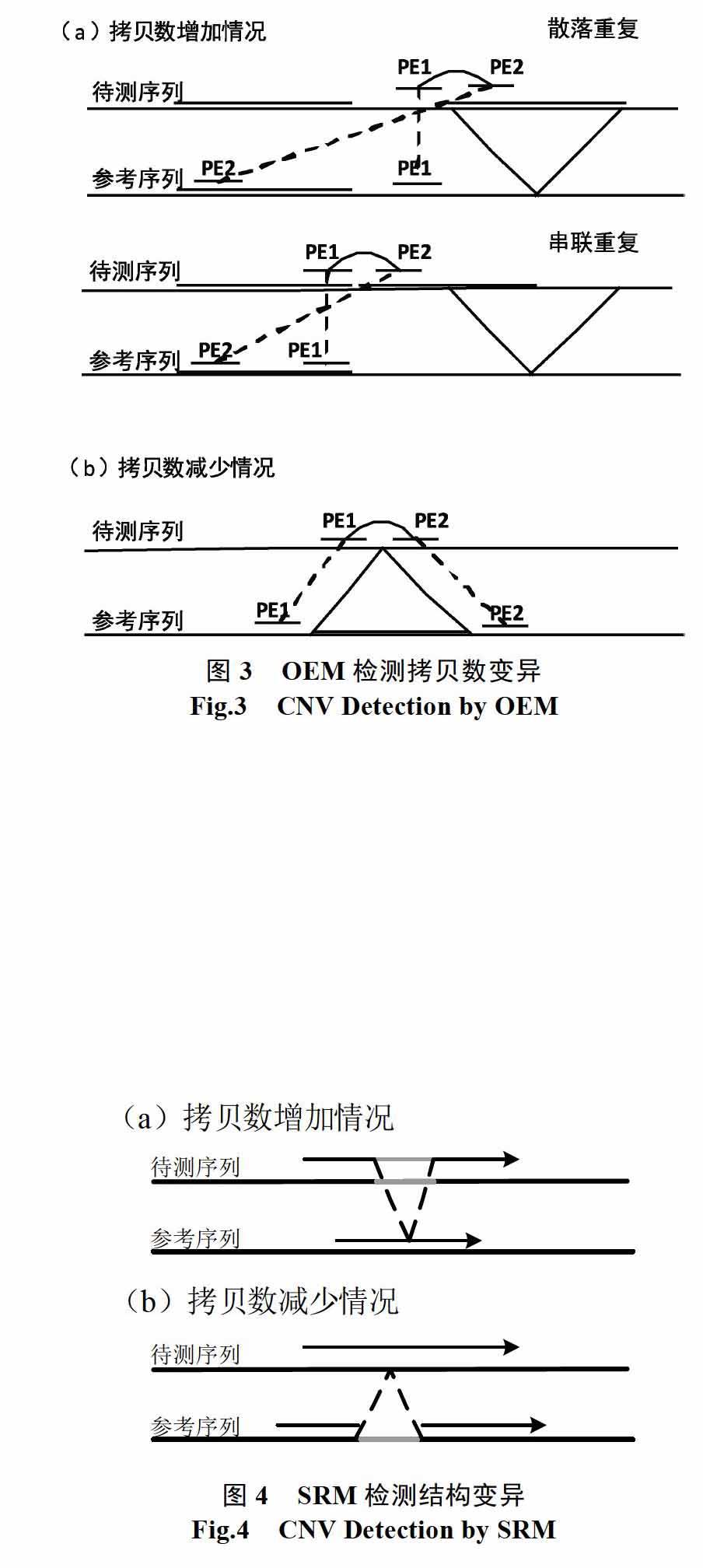
单端匹配(OEM)要求Paired-End Read的两端都能匹配到参考序列上,由图4可知,在检测拷贝数变异时根据拷贝数增加和拷贝数减少时情况有所不同。为了方便讨论,这里做如下定义:Paired-End Read双端间的距离称为插入长度(insert length),所有的Read的插入长度满足正态分布,令均值为IL,标准差为σ。进行匹配时,令前端匹配至参考序列上的位置为P1,后端匹配至参考序列的位置为P2,拷贝数区域的估计长度为CNV_Len。
(1)拷贝数增加时,若为散落重复,跨越拷贝数边缘的Paired-End Read匹配到参考序列上,一端会匹配至拷贝数序列区域内,另一端则可能会匹配至正常区域。这里判断该情况的标准为:
1.2 SRM检测
当被检测序列中存在结构变异时,位于结构变异边缘的读数据与参考序列无法整体匹配,但该读数据的前缀或者后缀则可以。而CNV属于结构变异,拷贝数增加本质上是一种插入变异,而拷贝数减少则是删除变异。SRM的核心思想是通过将原有的读数据分裂,将前缀和后缀匹配至参考序列,根据这些前后缀的匹配信息完成拷贝数变异检测的优化[15],图3是裂读法检测结构变异中插入和删除的示意图。本算法通过读数据分裂,根据前后缀的匹配信息验证现有工具检测得到的CNV,对于信号强度较弱的读计数异常进行过滤,从而提高检测精度。
为了方便描述,这里定义读数据Read的读长为RL,前缀为Readpre,长度为PL,PL=(RL-IL)/2,IL是可变参数,为Read中间部分忽略子串的长度,本方法中IL=RL/5,读数据的后缀为Readsuf,长度为SL,SL=PL。对于拷贝数增加,CNV边界将出现Readpre能匹配至参考序列非CNV所在区域而Readsuf能匹配至CNV区域的情况,而对于拷贝数的减少,则出现前后缀都匹配至非CNV所在区域的情况。基于以上分析,本文将非OEM的丢弃读数据分别取其PL长度的前后缀与参考序列进行比对,获得匹配坐标,对于多点匹配的情况采用加权随机分配方法,然后记录匹配坐标。
1.3 聚类分析与候选CNV的筛选
为了实现OEM和SRM检测获得的匹配坐标对候选基因的筛选,首先将上面记录的坐标信息进行聚类,本文采用的聚类方法为k均值聚类,由于OEM和SRM都是基于CNV边界附近的read信息来完成的,故k均值聚类的聚类中心数量k设置为候选CNV数量的两倍,分别对应于候选CNV的两个边界,且初始聚类中心指定为相应的边界坐标。OEM记录的两个匹配坐标仅使用其中靠近聚类中心的一个实施聚类。聚类分析结束后对每个聚类包含的点进行計数,表示为该聚类的置信度。
本文选用CNVnator作为产生候选CNV的工具,为了提高CNV检测的灵敏度,在候选CNV检测阶段,降低读深信号(RD signal)的阈值以检测出更多的候选CNV。然后根据候选CNV的质量值e-val结合聚类置信度进行最终CNV的筛选,筛选原则是:对于质量值较高的CNV直接保留,而质量值较低的CNV则进一步考虑相应的聚类置信度,置信度较高的也保留作为最终CNV,否则丢弃,不纳入最终结果。
2 实验验证与分析
为了测试本算法的有效性,本文同时采用模拟数据和真实的测序数据进行验证。采用模拟数据可以定制在序列中注入的拷贝数变异所在的位置以及拷贝数量,能够对算法的检测性能进行量化分析和比较;采用真实测序数据能够有效地反映算法应用于实际数据的检测效果,更接近于实际应用。作为比较,使用CNV检测工具CNV-Seq和CNVnator与本文提出的优化方法进行比较,检测性能利用精确性(Precision)、灵敏性(Sensitivity)和F1分数这三个参数进行评估。精确性表示检测结果被判定为正确的数据占全部检测结果的百分比;灵敏性表示的是判定为正确的检测结果占对应实际全部的CNV的百分比;F1分数是精确性和灵敏性的综合指标。令TP表示真阳性(True Position),表示检测结果和实际全部的CNV数据都是正类。FP表示假阳性(False Position),表示工具的检测结果是正类但实际是负类。FN表示假阴性(False Negative),表示工具的检测结果是负类但实际结果是正类。三个指标的公式分别为:精确性Pre=TP/(TP+FP),灵敏性Sen=TP/(TP+FN),F1=2*Pre*Sen/(Pre+Sen)。
2.1 模拟数据验证
模拟的CNV数据根据真实测序数据中拷贝数变异数据的不同类型和不同长度的特点进行设计,一共设计了两种拷贝数变异子类型:INS,DEL,INS对应于拷贝数的增加而DEL对应的是拷贝数的减少。同时设计了2个不同的长度区间:500—1000bp,1000—10000bp。每种变异长度都设计100变异。参考序列是hg19的11号染色体,模拟CNV数据使用的工具是Svsim (https://github.com/GregoryFaust/ SVsim),对该工具进行改写,生成包含准确变异信息的序列文件,以fasta格式进行存储。由变异序列文件再利用ART[16]生成Paired-End读文件,读长为100bp,以FASTQ格式进行存储。本文共生成三种测序深度的读数据,分别为30X,50X和70X。
本文选用的用于实验验证的工具为CNV-Seq和CNVnator与本文提出的优化方法进行比较,由于本文方法基于CNVnator,简称为CNVNOP,实验结果如表1所示。
由表1实验结果可以看出,无论是拷贝数增加和拷贝数减少的情况,CNVNOP方法比CNVnator和CNVSeq在精确性、灵敏度和F1值都有较好的表现,而当测序深度增大时,相应工具的检测性能都有所提升,相对而言50X的测序深度较30X的检测精度提高更明显一些。本文方法首先降低了CNVnator的信号筛选阈值已获得更多的候选结果,然后通过ORM和SRM进行筛选,实验结果表明该方法能够得到更高的检测性能。
2.2 真实数据验证
真实数据采用千人基因组项目中具有较高覆盖度的样本NA19240,这里仅取第1、2、10、11、20和22号染色体进行检测,从DVG数据库中查询可知这6条染色体的INS共1330个,DEL共777个。比较三种工具后的实验结果表2。由表2可以得到与模拟实验相同的的结论,优化后的方法具有较好的CNV检测性能。
3 總结与展望
本文提出了一种利用比对过程中丢弃的读数据对拷贝数检测进行优化的方法,以CNVnator为基础产生候选拷贝数,基于SRM和OEM的聚类结果进行筛选,有效地提高了检测精度和灵敏性,模拟数据和真实数据实验的结果验证了该方法的有效性。本文方法还具有两个重要的潜在优点:第一,它具有较好的通用性,本文方法中使用的CNVnator可以使用其它方法替代,例如:Speedseq,ReadDepth等。随着技术的进步,可能会有更高检测性能的方法,同样可以进行候选CNV检测方法的替换然后采用本文的优化方法;第二,通过本文方法可能计算出拷贝数变异的软切位点的位置,由OEM和SRM技术原理可知,其匹配位置通常会有两个,其中一个位于拷贝数变异区域,另一个则是增加的拷贝数区域位置,这对下游的功能分析能提供较好的帮助。本文方法也存在着一些不足之处,由于裂读匹配时需要将单个Read进行分割,因此要求的读长不能太小,否则匹配时非常容易产生多位置匹配,降低算法的检测精度;另外由于OEM利用了配对末端的insert size的信息,对于一些测序仪产生的非paired-end Read数据,本文方法也无法处理。
本文的优化方法能够有效提高检测精度,但还有值得完善的地方,首先本文采用的候选拷贝数的工具只采用一种工具进行检测,实际上这里可以多使用几种方法同时进行,利用群体优势来获得高可靠性的候选变异;其次在额外信息的使用中,除了OEM和SRM外,还有一些技术也可以被引入进行优化,例如:local assembly方法,在拷贝数变异区域附近进行局部拼接有助于获得进一步的变异细节,也能够提高检测的精度。这些方法将在纳入将来的研究中,进一步提高检测性能。
参考文献
[1] McCarroll, S. A., Extending genome-wide association studies to copy-number variation[J]. Hum Mol Genet, 2008. 17(R2): p. R135-42.
[2] Hinds, D. A., et al., Common deletions and SNPs are in linkage disequilibrium in the human genome[J]. Nat Genet, 2006. 38(1): p82-5.
[3] Redon, R., et al., Global variation in copy number in the human genome[J]. Nature, 2006. 444(7118): p. 444-54.
[4] Wong, K. K., et al., A comprehensive analysis of common copy-number variations in the human genome[J]. Am J Hum Genet, 2007. 80(1): p. 91-104.
[5] Bochukova, E. G., et al., Large, rare chromosomal deletions associated with severe early-onset obesity[J]. Nature, 2010. 463(7281): p. 666-70.
[6] Diskin, S. J., et al., Copy number variation at 1q21. 1 associated with neuroblastoma[J]. Nature, 2009. 459(7249): p. 987-91.
[7] Fanciulli, M., et al., FCGR3B copy number variation is associated with susceptibility to systemic, but not organ- specific, autoimmunity[J]. Nat Genet, 2007. 39(6): p. 721-3.
[8] Stefansson, H., et al., Large recurrent microdeletions associated with schizophrenia[J]. Nature, 2008. 455(7210): p. 232-6.
[9] McKernan, K. J., et al., Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding[J]. Genome Res, 2009. 19(9): p. 1527-41.
[10] Miller, C. A., et al., ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads[J]. PLoS One, 2011. 6(1): p. e16327.
[11] Li, J., et al., CONTRA: copy number analysis for targeted resequencing[J]. Bioinformatics, 2012. 28(10): p. 1307-13.
[12] Abyzov, A., et al., CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing[J]. Genome Res, 2011. 21(6): p. 974-84.
[13] H., L., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM[J]. eprint arXiv: 1303. 3997, 2013.
[14] Abyzov, A., et al., CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing[J]. Genome Res, 2011. 21(6): p. 974-84.
[15] Wang, J., et al., CREST maps somatic structural variation in cancer genomes with base-pair resolution[J]. Nat Methods, 2011. 8(8): p. 652-4.
[16] Huang, W., et al., ART: a next-generation sequencing read simulator[J]. Bioinformatics, 2012. 28(4): p. 593-4.
