
单肿瘤组织微卫星不稳定探测方法①
2019-09-24 06:20:16尚秋明韩鑫胤李瑞琳何小雨祝海栋牛北方
计算机系统应用 2019年9期
赵 丹,尚秋明,韩鑫胤,李瑞琳,何小雨,祝海栋,牛北方
1(中国科学院 计算机网络信息中心,北京 100190)
2(中国科学院大学,北京 100190)
3(中国互联网络信息中心,北京 100190)
近些年,基因组测序技术飞速发展,其发展经历包含三个阶段:第一代主要通过凝胶电泳和放射自显影技术确定待测的DNA 序列,其单次测序读长长、准确率高,但测序过程缓慢、吞吐量低;第二代核心思想为边合成边测序,即通过捕捉新合成的末端的标记来确定DNA 的序列,其特点是读长短、测序速度快、吞吐量大;第三代是单分子测序,成本较高,且目前测序技术尚未成熟[1].高通量测序技术作为第二代测序技术,满足了生命科学诸多领域的大规模测序需求,尤其在MSI 探测领域中,对于肿瘤的早期诊断、化疗敏感性判断[2]以及高危人群的圈定[3]等研究发挥了重要作用.
另外,作为肿瘤遗传不稳定的敏感指标,MSI 探测在化学治疗剂的选择[4,5]、预后判断[6,7]及家族性癌症风险评估[8,9]等方面均具有重要的临床意义.有效地利用MSI 探测结果可达到减少医疗盲目性,提高MSI 患者的治愈率,实现精准医疗的目标.目前,MSI 探测主要分为生物学实验方法和基于高通量测序数据的计算方法.通过分析高通量测序数据的MSI 探测方法,无需额外的临床测试或患者样本处理,大大减少由于实验方法依赖少量微卫星位点的探测或错配修复蛋白是否表达的局限性带来的误差与人力检测成本,因此被广大研究者所采用.但是,随着测序成本的急剧下降,测序通量呈指数提高,众多探测算法软件已无法满足使用者在运行速度、存储空间及用户体验等多方面的需求.基于计算的MSI 探测方法或软件主要存在两个限制条件:(1) 当使用样本的肿瘤与正常组织成对测序数据探测MSI 时,需要正常组织测序数据作为输入参照;(2) 仅使用肿瘤组织测序数据时,使用者需要针对不同的癌种或不同的测序平台得到的测序数据,分别使用大量MSS 样本的正常组织测序数据构建基准线,这不仅会在一定程度上造成特异性MSI 位点的丢失,而且给使用者带来了大量的额外工作,对存储空间和运行时间要求也非常高,无形之中给使用者提出了一个难题.因此,构建基于单肿瘤组织测序数据探测样本MSI 状态的模型十分必要.
本文主要分为四个部分.第1 部分,介绍已有的相关研究工作;第2 部分,在前期MSI 探测领域的研究基础上,针对MSIsensor1.1[10]无法实现只基于样本肿瘤组织测序数据探测MSI 状态的问题,提出一种采用信息熵理论的MSI 探测模型;第3 部分,对本文提出的模型进行性能评估测试;第4 部分,总结本文提出的模型的意义和展望未来MSI 探测研究方向.
1 相关工作
微卫星,又称为短串联重复序列(short tandem repeats,STRs),由长度在1~6 碱基对(base pair,bp)之间,重复次数为10~60 次的重复短核苷酸序列组成,其在真核生物的基因组中广泛存在[11].MSI 是指在DNA 复制期间,由于错配修复系统的缺陷(Mismatch Repair Deficiency,MMRD),导致重复短核苷酸序列的插入或缺失,出现微卫星新等位基因的现象.根据微卫星不稳定程度,MSI 分为MSI-H (Microsatellite Instability High),MSI-L (Microsatellite Instability Low)以及MSS,由于MSI-L 与MSS 没有显著性差异,本研究将MSI-L 视为MSS 处理.
随着下一代测序技术的发展,基于高通量测序数据的MSI 检测方法及软件逐渐涌现.目前,无需输入肿瘤与正常组织成对测序的方法和软件有mSINGS[12]、Lu 等人提出的基于插入和缺失变异(Insertion and Deletion,Indel)的MSI 检测方法[13]、MSIseq[14]和MSIpred[15].对BAM (Binary sequence Alignment/Map format)文件直接进行分析的mSINGS 无需配对的正常组织测序数据做参照,但在对样本进行分析之前,需要足够的MSS样本正常组织测序数据建立各个位点的基准线,根据3σ法则判断各个位点的稳定性,最后通过统计不稳定位点的比例获得样本的MSI 状态.Lu 等人提出的方法使用PI/PD 作为判别样本MSI 状态的指标,并通过实验证实该方法的可行性.其中,PD 是指微卫星区域Deletion 占所有Deletion 的比例,PI 是指微卫星区域Insertion 占所有Insertion 的比例.MSIseq 基于决策树算法的分类器在单核苷酸替代(Single Nucleotide Substitution,SNS)和Indel 短突变的高级别数据上进行模型训练和测试,该方法在测试时仅使用微卫星区域小片段碱基插入与删除的比例这一特征.MSIpred 利用支持向量机对从突变数据中提取的12 个特征进行模型训练和测试.MSIseq、MSIpred 等是从突变注释格式(Mutation Annotation Format,MAF)中提取特征用于模型训练和测试,但MAF 是由肿瘤与配对正常组织组织的测序数据产生.因此,提供一个直接对肿瘤组织测序数据的BAM 文件进行分析的检测工具十分必要.
基于肿瘤与正常组织成对测序数据的MSIsensor 1.1和MANTIS[16]可以直接对BAM 文件进行分析.MANTIS将样本的肿瘤组织与配对的正常组织在每个微卫星位点的等位基因分布看作两个向量,定义这两个向量的L1范数作为衡量该位点稳定程度的度量,最后通过统计该样本所有位点的L1范数的平均值获得该样本的MSI 分数.MSIsensor 通过卡方检验比较肿瘤组织和配对的正常组织的相同微卫星位点的等位基因分布是否显著不同判断该位点是否稳定,最后统计不稳定的位点占总位点的比例判断样本的MSI 状态[17,18].
通过对MSIsensor1.1和MANTIS 软件之间的对比,本研究发现MSIsensor1.1 具有以下两方面优点:(1)设计基于C++实现、计算效率高;(2) 通过使用全基因组范围内的微卫星位点信息的探测和卡方检验方法判断微卫星状态,准确性非常高.因此,本研究选择该软件进行优化.
MSIsensor1.1 基于卡方检验的探测模型包含以下两个功能模块:
(1)获取位点基本信息模块.通过对参考基因组文件(Reference Genome.fa)执行扫描(scan)命令,构建一个由微卫星位点基本信息组成的序列字典(microsatellites.list),其基本信息包括:微卫星位点所在染色体号及其在染色体上的起始位置、微卫星重复单元的大小及重复次数以及微卫星位点的左右翼信息;
(2)探测样本MSI 状态模块.通过对肿瘤组织测序数据文件(Sample.tumor.bam)、正常组织测序数据文件(Sample.normal.bam)和microsatellites.list 执行msi 命令获得待测样本的MSI 分数(MSIscore).该模块共有四个结果文件,分别为:MSI 分数文件(Output 文件)、微卫星位点等位基因分布文件(Output_dis 文件)、germline 位点文件(Output_germline 文件)和somatic位点文件(Output_somatic 文件).该模块具体步骤如下:① 计算每个位点在样本肿瘤与正常组织成对测序数据上微卫星的长度分布,并筛选在肿瘤和正常组织测序数据中测序深度均大于20 的微卫星位点;② 利用卡方检验比较符合条件①的微卫星位点在肿瘤与正常组织中的分布差异,若显著不同,则判定该位点为不稳定的微卫星位点;③ 通过统计不稳定位点占总位点的比例获得样本的MSIscore,进而判断该样本的MSI状态.
2 实验方法
目前,基于肿瘤与正常组织成对测序数据的MSI探测方法,利用配对的正常组织测序数据为参考,对肿瘤组织的微卫星位点的不稳定情况进行评判,最终达到判断样本MSI 状态的目标.对于缺少配对的正常组织测序数据做参照的情况,为了避免使用大量MSS 样本的正常组织测序数据构建基准线而丢失特异性MSI 位点,本文提出一种新模型,该模型根据肿瘤组织每个微卫星位点等位基因分布的混乱情况,来判断该位点及样本的MSI 状态.
在正常细胞中,错配修复系统(Mismatch Repair,MMR)提供了一种高效的机制来纠正DNA 复制过程中发生的错误.当MMR 受损时,例如错配修复基因MLH1、MSH2和MSH3 的失活,将导致基因序列——特别是微卫星位点重复单元的错误插入或删除得不到纠正,微卫星位点的重复单元的重复次数出现波动,即MSI 发生[7].从酵母的研究中发现,在DNA 错配修复蛋白突变后,微卫星中短缺失的数量明显增加,而微卫星中插入的数量没有明显变化[8].后续研究也证实了,由于MMRD,微卫星区域会发生小片段碱基的插入与缺失.受已有研究成果的启发,我们考虑到,与MSS 样本相比,MSI 样本肿瘤组织的微卫星位点的重复单元的重复次数的分布会因为MMRD 呈现比较混乱的状态.图1为MSS 样本与MSI 样本的肿瘤组织在同一微卫星位点的等位基因分布.从图中可以看出,MSI 样本的微卫星位点等位基因分布与MSS 样本相比较混乱.鉴于MSI 样本肿瘤组织微卫星位点测序数据这一特性,本研究提出使用信息熵理论来探测样本的MSI 状态.

图1 MSI 样本与MSS 样本的同一微卫星位点等位基因分布比较
信息熵是描述“混乱”程度的量度.系统越有序,信息数据越集中的地方,熵值越小;系统越混乱,信息数据越分散的地方,熵值越大[19].扩展后的MSIsensor 工作流程如图2所示.

图2 MSIsensor 工作流程图
图中虚线所指是MSIsensor1.1 已有模块,实线所指功能是本文在实现探测样本MSI 状态功能中提出的基于信息熵理论在样本单肿瘤组织测序数据进行探测的新模型.该模型共有三个输出,分别为:Output 文件、Output_dis 文件、Output_somatic 文件.新模型主要包含以下子模块:
(1)目标位点筛选功能.通过计算每个微卫星位点在肿瘤组织测序数据上的分布,可筛选出测序深度大于20(默认)的微卫星位点.
(2)目标位点过滤功能.由于测序过程中可能会带来背景噪声信息,因此,对(1)筛选出的目标位点集进行过滤.过滤规则为:过滤掉肿瘤组织在该位点支持reads 数小于3 的等位基因(被认为是噪声数据),接下来以剩下的等位基因数作为样本在该位点的等位基因数.
(3)加权信息熵值计算功能.根据步骤(2)得到的每个位点等位基因分布情况,计算加权信息熵值.信息熵值计算公式如下:

其中,pi指的是针对位点的每个重复次数,其不为零支持reads 数占该位点支持reads 数总和的比例.支持reads 数不为零的重复次数的个数会较大程度影响每个位点的信息熵值,即每个位点的信息熵值会因支持reads 数不为零的重复次数的个数变化而跨越幅度较大.为了方便后续信息熵值阈值(cutoff)的确定,对每个位点的熵值赋予一定的权重(weight).若该位点的H(U)*weight大于等于0.3(默认),则判定该位点不稳定.其中,weight等于该位点支持reads 数不为零的重复次数的个数的倒数.在TCGA 数据集的不同癌种上使用不同的阈值进行多次实验,通过比较得到,当使用阈值0.3 时,在不同癌种上Accuracy 都比较高.图3给出了在TCGA 数据集上随着cutoff变化时的Accuracy 趋势.

图3 不同cutoff 下的Accuracy 比较
(4) MSIscore 计算功能.通过统计由(3)得到不稳定位点数占满足条件(1)的微卫星总位点数的比例,获得该样本的MSIscore,进而判断样本的MSI 状态.
3 实验结果与讨论
本研究使用的数据来自国际癌症基因组图谱(The Cancer Genome Atlas,TCGA)计划[20]、欧洲基因组-表型组档案(European Genome-phenome Archive,EGA)[21,22]和北京肿瘤医院(Beijing Cancer Hospital),包含的癌症类型有胃癌(STomach ADenocarcinoma,STAD)、子宫内膜癌(Uterine Corpus Endometrial Carcinoma,UCEC)、结肠癌(COlon ADenocarcinoma,COAD)和直肠腺癌(REctum ADenocarcinoma,READ),具体情况如表1所示.
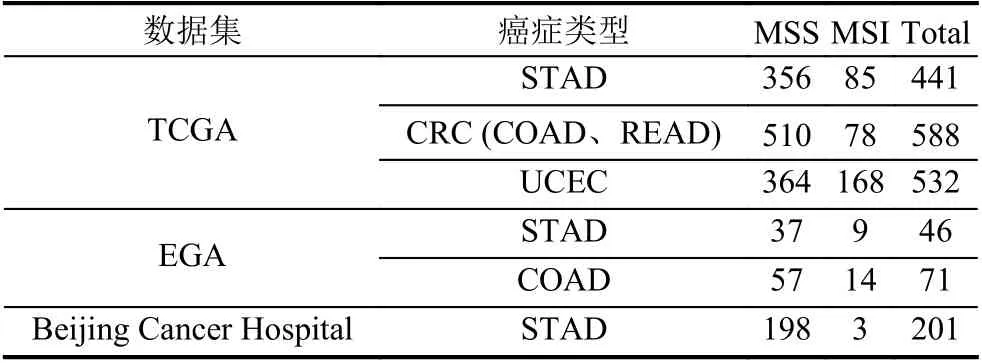
表1 TCGA、EGA和Beijing Cancer Hospital 数据集
为了衡量MSIsensor 扩增的使用单肿瘤组织测序数据探测MSI 模块的表现性能,本研究将其与MSI sensor 基于卡方检验的MSI 探测模块在表1所示数据上的测试结果进行对比.由于不同的癌症类型或不同测序平台得到的测序序列会稍有不同,本文不建议使用统一的MSIscore 阈值划分MSS 样本和MSI 样本.针对基于卡方检验在肿瘤与正常组织成对测序数据上探测MSI 的模块,本研究在TCGA STAD、TCGA CRC、TCGA UCEC、EGA STAD、EGA COAD和Beijing Cancer Hospital 数据集上采用的MSIscore 阈值分别为3、10、3、3、10、和15,其各项性能指标表现如表2所示,其中,准确率(Accuracy)表示预测状态正确的样本数占待检测样本总数的百分比;精确率(Precision)表示软件预测的真实MSI 样本数占软件预测出的MSI 样本总数的百分比;灵敏度(Sensitivity)表示软件预测的真实MSI 样本数占MSI 总样本数的百分比;特异性(Specificity)表示软件预测的真实MSS样本数占MSS 总样本数的百分比;F-分数(F-score)表示Sensitivity和Precision 两个性能度量的调和平均数,用来综合评估软件的性能.具体分类效果如图4所示.针对基于信息熵在单肿瘤组织上探测MSI 的模块,本研究在TCGA STAD、TCGA CRC、TCGA UCEC、EGA STAD、EGA COAD和Beijing Cancer Hospital数据集上采用的阈值分别为13、20、13.5、11、15和20.该模型的各项性能指标表现如表3所示,具体分类效果如图5所示.

表2 基于卡方检验探测模块的测试结果
以上结果证明,在TCGA 上确定的对每个位点判定是否稳定的加权信息熵阈值0.3,在EGA、Beijing Cancer Hospital 数据集上同样适用.同时,该结果也表明本文提出的基于信息熵理论在单肿瘤组织测序数据上探测样本MSI 状态的方法是可行的.
基于信息熵的MSI 探测方法只需要对单肿瘤组织测序数据进行分析,因此,在探测MSI 的前期准备工作中不需要对正常组织进行测序,可以节省测序成本;且与MSIsensor1.1 在肿瘤与正常组织测序数据上探测MSI 的方法相比较,其运行效率提高了一倍.
4 结论与展望
本研究针对样本单肿瘤组织的外显子或全基因组测序数据的MSI 探测问题,选择从高通量测序数据入手,基于已有软件MSIsensor1.1,增加采用信息熵理论探测MSI 状态的模块.扩增后的软件不仅可针对肿瘤与正常组织成对测序数据探测样本MSI 状态,而且当无配对的正常组织测序数据做输入参照物和不严重影响精度的情况下,可满足样本单肿瘤组织测序数据的MSI 探测需求,是一套功能完备、兼容多种测序数据模式、具备实用性和前瞻性的MSI 探测软件.
后续我们将围绕以下两方面工作展开:(1)基于此工作深入挖掘每个位点信息并筛选与肿瘤发生发展相关的基因,这样不但可以节省测序的成本,而且可以提高检测的正确率;(2)探究基于血浆的MSI 探测方法.癌细胞具有扩散性,当癌细胞侵犯血管进入血液时,血液中也会有MSI 信号,但此时信号很微弱,不易检测.目前大多数MSI 检测局限于组织测序数据,但是对于不方便提取组织的患者,开发基于血浆的MSI 探测方法具有重要的意义.下一步,我们将围绕此工作展开,探求更强、更符合实际应用需求的MSI 探测软件.

图4 基于卡方检验探测模块的测试结果

表3 基于信息熵理论探测模块的测试结果

图5 基于信息熵理论探测模块的测试结果
猜你喜欢
特产研究(2022年6期)2023-01-17 05:05:06
军民两用技术与产品(2022年1期)2022-06-01 06:28:50
智慧健康(2021年17期)2021-07-30 14:38:32
电子测试(2017年12期)2017-12-18 06:35:48
四川动物(2017年4期)2017-07-31 23:54:19
雷达学报(2017年6期)2017-03-26 07:52:58
现代检验医学杂志(2016年5期)2016-08-20 03:16:54
法医学杂志(2015年4期)2016-01-06 12:36:40
池州学院学报(2015年3期)2016-01-05 01:13:00
集美大学学报(自然科学版)(2015年4期)2015-02-28 01:13:37
