
水质丁基黄原酸测定难点探讨
2019-09-17 08:03:24王海燕
中国环境监测 2019年4期
夏 勇,王海燕,刘 鑫,林 武
攀枝花市环境监测中心站,四川 攀枝花 617000
黄药,即黄原酸盐,按其化学组成也可称为烃基二硫代碳酸盐,结构通式为RO-C-SSMe(R为烃基,Me为Na+或K+),一般由醇、苛性碱(氢氧化钠或氢氧化钾)及二硫化碳合成。因此,按照合成使用的苛性碱不同,黄药分为钾黄药和钠黄药两大类。根据合成使用的醇不同,黄药又有不同的品种,常见的有乙基、丙基、异丙基、正丁基、异丁基、戊基、异戊基黄药,这些黄药在国内均有生产和使用。黄药主要用作金属硫化物矿石的浮选收集剂,也常用于橡胶的硫化[1]。
黄药的环境效应不容忽视。黄药具有恶臭,硫化矿的选矿废水中即使只残存极少量的黄药,仍能使尾矿水及周围空气产生异臭。含有黄药的选矿废水大量排放时,还可使受纳水体变臭。在水中,黄药会抑制多种水生生物的生长,使鱼类饵料减少,生长缓慢。另外,黄药还对一些鱼类和蛙类具有致畸性和致死性[2]。如丁基黄原酸钠对草鱼胚胎具有强烈的致畸作用,畸形的主要症状为弯体和体表瘤状赘生物[3]。长期毒性试验表明,1 mg/L的戊基钾黄药可导致红鳟死亡[4]。根据浮选药剂对鱼和水蚤类的毒性排名[5],丁基黄药、异戊基黄药属于中等毒性物质,乙基黄药属于强毒性物质。黄药对动物和人的危害主要表现为使神经系统和肝脏器官受害,且其在微酸条件下能分解出具有神经毒性的二硫化碳,二硫化碳可通过血液进入大脑,使神经系统产生病症,能对造血系统产生不良影响,还会引起肌肉痛、情绪不稳定、食欲不振、高血压等[6]。
丁基黄药的水解产物丁基黄原酸被中国《地表水环境质量标准》(GB 3838—2002)列为集中式生活饮用水地表水源地水质特定监测项目,限值为0.005 mg/L;也被《生活饮用水卫生标准》(GB 5749—2006)列为生活饮用水水质参考指标,限值为0.001 mg/L。然而,丁基黄药仅是黄药中的一种,其他黄药的环境危害也不容忽视。目前,水质丁基黄原酸分析过程中存在丁基黄原酸及其盐概念混淆,工业品中共存黄药干扰丁基黄原酸测定,样品采集及保存阶段目标物不稳定易分解,部分分析方法前处理阶段目标物易损失,一些分析方法选择性不好等技术难点。笔者结合实验对水质丁基黄原酸测定中容易出现的问题及应该注意的事项进行介绍,以期为水质丁基黄原酸的准确测定提供参考。
1 概念混淆
概念的混淆主要表现在混淆丁基黄原酸及其盐。丁基黄原酸是丁基黄药的水解产物,被《地表水环境质量标准》(GB 3838—2002)和《生活饮用水卫生标准》(GB 5749—2006)纳入了监测范畴。但这2个国家标准中并没有明确丁基黄原酸以何种分子式表示。《生活饮用水标准检验方法 有机物指标》(GB/T 5750.8—2006)中丁基黄原酸标液的配制叙述如下:“丁基黄原酸标准储备溶液〔ρ(C4H9OCSSH)=100 μg/mL〕:称取0.027 8 g丁基黄原酸钾(C4H9OCSSK,含量为90%),置于250 mL容量瓶内,加3滴氢氧化钠溶液,用纯水溶解,定容”。可见,这里的配制过程明确指出丁基黄原酸是以C4H9OCSSH计。按照该过程配制出来的丁基黄原酸钾(C4H9OCSSK)浓度为100 μg/mL,而通过折算丁基黄原酸(C4H9OCSSH)的实际浓度为79.8 μg/mL。这里便混淆了丁基黄原酸及其盐。后面出台的几个行业标准分析方法[7-9]也明确丁基黄原酸是以C4H9OCSSH计,在配制过程中区分了丁基黄原酸及其盐,并考虑了两者之间的换算关系。因此,在使用《生活饮用水标准检验方法 有机物指标》(GB/T 5750.8—2006)测定丁基黄原酸时要注意丁基黄原酸标准储备液的配制问题。另外,建议《地表水环境质量标准》(GB 3838—2002)和《生活饮用水卫生标准》(GB 5749—2006)中明确丁基黄原酸以C4H9OCSSH计。
2 样品采集和保存
丁基黄原酸不稳定,有效保存样品是丁基黄原酸监测的重要环节之一。目前,USEPA、ISO、欧盟和日本的有关质量标准和排放标准中,没有丁基黄原酸这一控制指标,也没有查到相关标准分析方法。中国的丁基黄原酸标准分析方法详见表1。

表1 国内标准分析方法中有关丁基黄原酸测定的水样采集和保存规定Table 1 Regulations about sample collection and preservation in the determination of butylxanthic acid in water according to the national standard methods
《生活饮用水标准检验方法 有机物指标》(GB/T 5750.8—2006)并未指明采样瓶材质、保存条件和保存时间。后来颁布的几个行业标准分析方法[7-9]都逐步完善,明确了样品采集和保存等细节。几个行业标准不一致的地方主要集中在采集到的水样酸碱度调节到什么程度以及样品保存期。仅在调节pH、避光冷藏保存条件下,样品在保存期上的差异可能是由于考察保存期的样品供试浓度高低差异造成的。对于一个未知样品,在无法判断其丁基黄原酸浓度大小的情况下,建议采集后尽快分析,最长不超过1 d。针对样品保存酸碱度问题,包括《水质 丁基黄原酸的测定 吹扫捕集/气相色谱-质谱法》(HJ 896—2017)以及《水质 丁基黄原酸的测定 液相色谱-三重四极杆串联质谱法》(HJ 1002—2018)在内的标准和文献[1,10]中都表明弱碱性环境更有利于丁基黄原酸的保存。
3 样品前处理
《水质 丁基黄原酸的测定 紫外分光光度法》(HJ 756—2015)样品前处理过程中明确指出,采集的待测样品混浊或者含有不溶物质,需过孔径为0.45 μm滤膜,但并未指出使用什么材质的滤膜。有研究[11-12]表明,对于有机物分析,滤膜材质的选择极其重要,选择不当将会导致分析结果呈假阳(阴)性。朱红霞等[13]考察了混合纤维、聚醚砜、尼龙和聚四氟乙烯(PTFE)4种材质滤膜对丁基黄原酸回收率的影响,结果表明,混合纤维、聚醚砜和PTFE这3种材质的滤膜对丁基黄原酸的测定影响不大。《水质 丁基黄原酸的测定 液相色谱-三重四极杆串联质谱法》(HJ 1002—2018)[9]起草单位曾考察了聚醚砜、尼龙、玻璃纤维、亲水性聚丙烯和PTFE滤膜对丁基黄原酸测定的影响,结果表明,玻璃纤维滤膜、亲水性聚丙烯滤膜和PTFE滤膜待测物通过率为90%~110%,聚醚砜滤膜待测物通过率为70%~80%,而尼龙滤膜待测物通过率为10%左右,因此推荐选择孔径为0.22 μm的玻璃纤维滤膜、亲水性聚丙烯滤膜和PTFE滤膜过滤样品。综合来看,前述2个研究均表明PTFE材质滤膜对丁基黄原酸分析影响小,应成为过滤的首选材质。
4 物质纯度
物质的纯度是准确定量分析的基础。黄原酸盐合成所需的醇中可能含有其他杂质醇,最终会导致合成的这种黄原酸盐中存在杂质黄原酸盐。BARNES等[14]考察了几种商品化黄原酸盐的纯度,见图1。
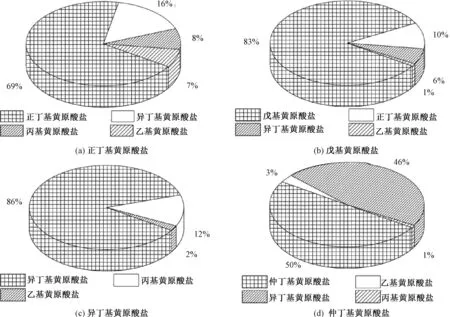
图1 部分商品化黄原酸盐及其杂质[14]Fig.1 Impurities in some commercially available butylxanthates[14]
由图1可见,单一品种的工业级黄原酸盐中仍然含有少量其他共存黄原酸盐。如所考察的正丁基黄原酸盐实际含量为69%,共存有16%的异丁基黄原酸盐,8%的丙基黄原酸盐和7%的乙基黄原酸盐。笔者利用超高效液相色谱三重四极杆串联质谱仪对国内某选矿药剂厂生产的工业级丁基黄药水解产物及其共存黄原酸进行定性分析(图2),并与几种烃基黄原酸的标准谱图(图3)进行比较。

图2 工业丁基黄药中烃基黄原酸的总离子流Fig.2 TIC chromatograms of alkyl xanthic acid in commercially butyl xanthate
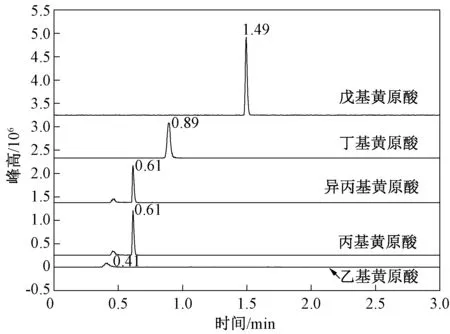
图3 烃基黄原酸标准液的总离子流Fig.3 TIC chromatograms of alkyl xanthic acid standard samples
实验所用分离条件如下:色谱柱为Waters ACQUITY UPLC BEH C18(50 mm×2.1 mm,1.7 μm),流动相A为乙腈,流动相B为氨水溶液(pH约为9.5),初始比例为5%的A,保持0.5 min,1 min内升高到95%,保持0.5 min,0.5 min内降为5%,保持1 min,流速为0.3 mL/min,柱温为35 ℃。实验结果表明,该工业级丁基黄药水解产物中未检测到乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸。然而,在丁基黄原酸(保留时间为0.90 min)的附近出现一个峰,保留时间为0.98 min(图2),该峰是采用丁基黄原酸的质谱分析参数获得,并且保留时间与丁基黄原酸保留时间(0.90 min)非常接近。笔者利用质谱仪分析丙基黄原酸和异丙基黄原酸母离子和子离子的时候,发现两者具有相同的定性定量离子对。据此推测该工业品中丁基黄原酸附近出现的峰可能是其同分异构体(如异丁基黄原酸)的峰。因此,水质丁基黄原酸的监测需要选择性好的分析方法。
5 丁基黄原酸常用分析方法
5.1 铜试剂亚铜分光光度法
铜试剂亚铜分光光度法是《生活饮用水标准检验方法 有机物指标》(GB/T 5750.8—2006)及《地表水环境质量标准》(GB 3838—2002)中指定的分析方法。其原理是在pH为5.2的盐酸羟胺还原体系中,将铜离子还原成亚铜离子。水中的丁基黄原酸与亚铜离子生成黄原酸亚铜后被环己烷萃取。黄原酸亚铜再与铜试剂作用,生成橙黄色铜试剂亚铜,于436 nm处定量测定。笔者通过实验考察了该方法的选择性。具体是分别以乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸代替丁基黄原酸,并按照铜试剂亚铜分光光度法进行分析,最后利用紫外可见分光光度计在350~500 nm波长范围内扫描最终产物的吸收光谱,如图4所示。

图4 铜试剂亚铜法所得终产物的吸收光谱Fig.4 UV-Vis spectra of end-products formed by sodium diethyldithiocarbamate cuprous method
结果表明,除乙基黄原酸和丙基黄原酸外,异丙基黄原酸和戊基黄原酸按照铜试剂亚铜分光光度法所得终产物与丁基黄原酸按照铜试剂亚铜分光光度法所得终产物最大吸收波长均在436 nm处。由此可见,利用铜试剂亚铜分光光度法测定丁基黄原酸时,异丙基黄原酸和戊基黄原酸将会干扰测定。
5.2 紫外分光光度法
丁基黄原酸在波长为301 nm处有最大吸收峰。当pH<2时,丁基黄原酸完全分解,同时吸收峰消失。通过酸分解前后吸光度之间的差值可以得出丁基黄原酸的浓度。目前,该法也有相应的环境保护标准分析方法出台[7]。笔者通过实验表明,丁基黄原酸、乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸均在301 nm处有最大吸收峰,且酸分解后该吸收峰均消失,见图5。因此,乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸会干扰丁基黄原酸的测定。此外,贺心然等[15]研究表明,亚硫酸盐、连二硫酸盐、偏亚硫酸氢盐在301 nm处没有吸收峰,但是在加酸酸化后生成的二氧化硫在301 nm处有吸收峰,也给测定带来干扰。
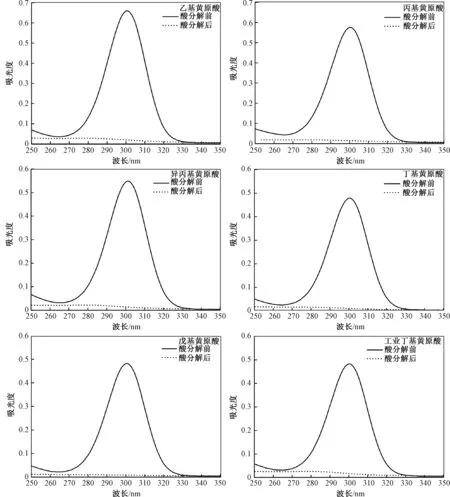
图5 烃基黄原酸分解前后紫外吸收光谱Fig.5 UV spectra of alkyl xanthic acid before and after decomposition by acid
5.3 气相色谱法/气相色谱质谱法
水中丁基黄原酸在酸性条件下分解产生CS2,通过气相色谱或气相色谱质谱测定CS2可间接测定丁基黄原酸含量。栗颖明等[16]率先利用该性质建立了顶部空间气相色谱测定水中丁基黄原酸的方法。该法是以注射器为反应器,将一定量的水样注入该注射器,再加入硫酸,接着抽吸一定体积空气形成顶部空间,用堵头密封该注射器,摇匀后70 ℃水浴加热1.5 h,最后吸取该注射器顶部空间的部分平衡蒸汽注入气相色谱,火焰光度检测器检测,最低检测浓度为0.3 μg/L。杨丽莉等[17]利用顶空-气相色谱-质谱联用仪间接测定了水中丁基黄原酸。与传统的分光光度法相比,该法简便、快捷,丁基黄原酸在0.010~1.00 mg/L范围内线性良好,样品加标回收率为95.1%~100.6%,相对标准偏差为3.1%~6.5%,方法检出限为0.002 mg/L。该课题组[18]也利用吹扫捕集/气相色谱-质谱联用仪间接测定了丁基黄原酸,该法在0.25~10.0 μg/L范围内线性良好,样品加标回收率为98.3%~105%,相对标准偏差为5.92%~10.7%,方法检出限为0.07 μg/L,比顶空法灵敏度更高。然而,烃基黄原酸酸分解后均能产生CS2,因此,利用CS2间接测定是不能区分烃基黄原酸的。另外,《水质 丁基黄原酸的测定 吹扫捕集/气相色谱-质谱法》(HJ 896—2017)指出,吹扫捕集/气相色谱-质谱法不适合含盐量高于25 g/L的高盐工业废水及存在代森锌(锰)类农药或二乙基二硫代氨基甲酸盐(如铜试剂)等物质水样丁基黄原酸的测定。
综上,当水样检出丁基黄原酸时,铜试剂亚铜分光光度法、紫外分光光度法、气相色谱/气相色谱质谱间接测定法在选择性方面均难以满足丁基黄原酸准确测定的要求,还需采用其他方法进一步确认。
5.4 离子色谱法
目前报道的测定丁基黄原酸的离子色谱法主要有2种方式,一种是离子色谱电导检测法,一种是离子色谱紫外检测法。由于《地表水环境质量标准》(GB 3838—2002)和《生活饮用水卫生标准》(GB 5749—2006)中对丁基黄原酸的限值较为严格。因此,在采用离子色谱法测定水中丁基黄原酸时通常要采取大体积进样方式来降低方法检出限。方黎等[10]以IonPac AS20柱为分离柱,30 mmol/L NaOH为淋洗液,1.0 mL/min流速,电导检测器检测丁基黄原酸。当进样1 mL时,丁基黄原酸在5~200 μg/L浓度范围呈良好线性关系,方法检出限为2.0 μg/L。李仁勇[19]采用IonPac AS16柱为分离柱,125 mmol/L NaOH为淋洗液,0.5 mL/min流速,301 nm紫外检测丁基黄原酸。当进样500 μL时,丁基黄原酸在0.5~1 000 μg/L宽浓度范围呈良好线性关系,方法检出限为0.1 μg/L,达到或者优于一些色谱质谱法[9,17]的检出限,而且在8 min内就可以完成分离,常见共存阴离子并不干扰测定。然而,这些方法中都没有考察其他黄原酸的干扰问题。朱红霞等[20]以IonPac AS20柱为分离柱,采用多步梯度淋洗方式分离了水中乙基黄原酸钾、异丙基黄原酸钾、丁基黄原酸钾、异丁基黄原酸钾、戊基黄原酸钾和异戊基黄原酸钾6种黄原酸盐。进样500 μL时,丁基黄原酸盐的检出限为0.003 mg/L。但该法中需要淋洗液发生器能支持多步梯度,而一些水质监测部门配备的离子色谱仪淋洗液发生器并不支持多步梯度淋洗。笔者尝试采用等度淋洗方式对常见烃基黄药进行分离,具体是以IonPac AS20柱为分离柱,40 mmol/L KOH等度淋洗,1.0 mL/min流速,电导检测,色谱图见图6。结果表明,所选实验条件下,常见阴离子(氟离子、氯离子、硫酸根、硝酸根、磷酸根),乙基黄原酸,丙基黄原酸,异丙基黄原酸,戊基黄原酸并不干扰丁基黄原酸的测定。特别需要说明的是,由于实验条件限制,并没有考察异丁基黄原酸和异戊基黄原酸对丁基黄原酸的干扰。
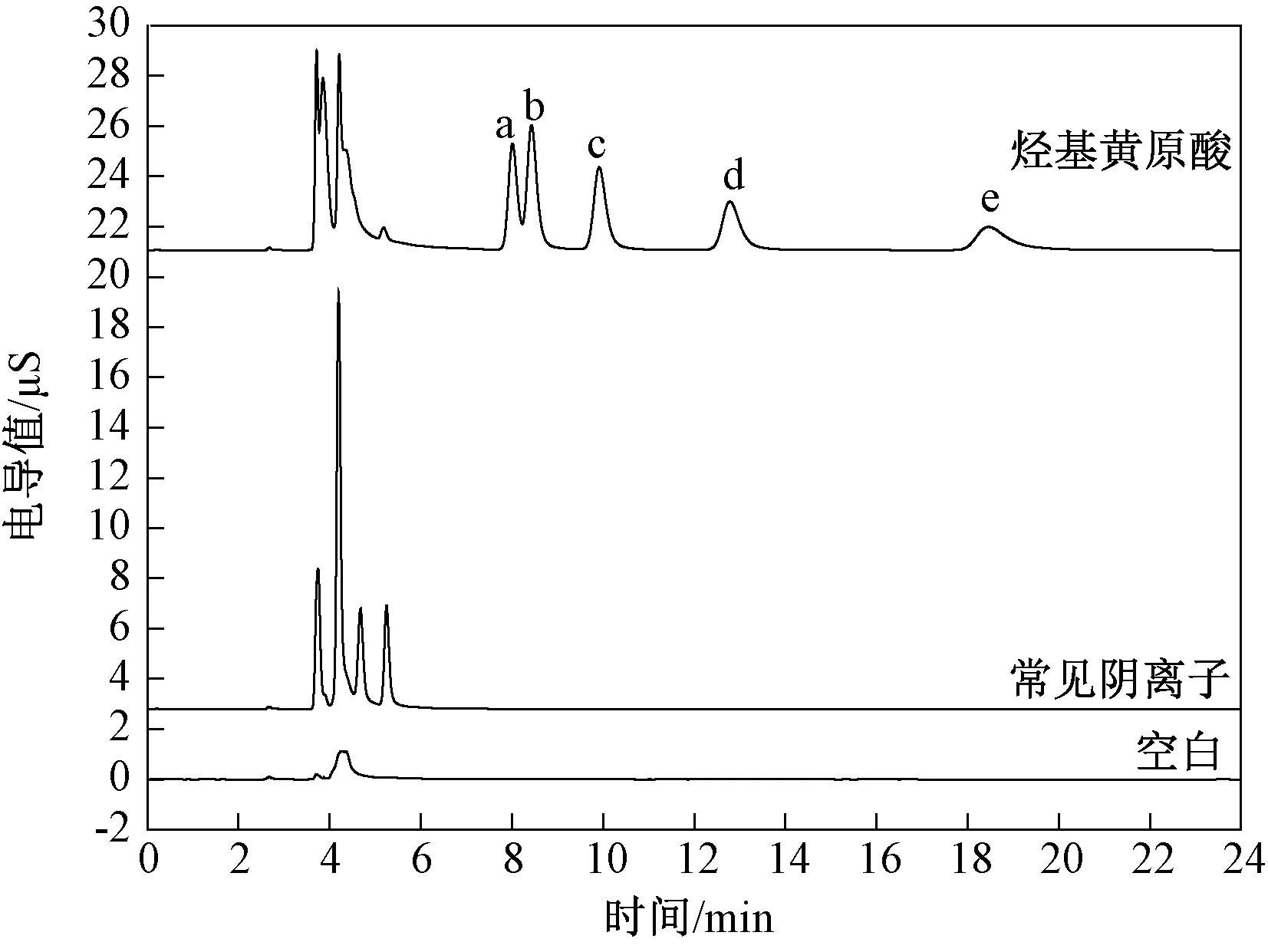
注:峰a.异丙基黄原酸;峰b.乙基黄原酸;峰c.丙基黄原酸;峰d.丁基黄原酸;峰e.戊基黄原酸。图6 烃基黄原酸离子色谱Fig.6 Ion chromatograms of alkyl xanthic acid
5.5 液相色谱法
黄原酸根极性较强,在非极性色谱柱上保留较弱,有可能会受基质的干扰。为解决黄原酸根保留弱的问题,ZHOU等[21]采取柱前衍生的方法将其转化成双黄原酸或者金属络合物,使其电荷部分屏蔽,极性减弱,疏水性增强,这些分子在常见的反相固定相上就有适中的吸附力。同时,由于分子基团和分子间碳原子数差别增大而有利于色谱分离。但研究表明,无论是借助双黄原酸或金属络化物,所得到的不只是这些黄原酸各自形成的、碳链对称的色谱峰,而且伴有交错形成的非对称碳链的色谱峰,致使色谱图变得很复杂,需要复杂的计算过程进行分析。近年来,也有利用反相液相色谱直接进样分析丁基黄原酸的报道。彭涛等[22]采用直接进样-超高效液相色谱分离-紫外吸收检测方式测定了地表水中丁基黄原酸。该法以0.050 mol/L乙酸铵溶液(pH用氨水调至约9.5)∶乙腈=80∶20等度洗脱,Waters ACQUITY UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7 μm)分离,301 nm为检测波长,进样10 μL时,丁基黄原酸在0.5~20.0 μg/L浓度范围内线性良好,方法检出限为0.8 μg/L,2 min内就可以完成一个样品分析。张凌云[23]以Agilent ZONREAX Bonus-RP(150 mm×4.6 mm,5 μm)为液相色谱柱,采取与文献[22]同样的流动相和检测波长,进样30 μL时,丁基黄原酸在2~100 μg/L浓度范围内线性良好,方法检出限为0.7 μg/L。图5表明,烃基黄原酸在301 nm处均有最大吸收波长,而前述2个直接进样液相色谱分析方法并没有讨论在所选色谱分离条件下其他烃基黄原酸对丁基黄原酸分析的干扰。笔者采用文献[22]报道的色谱分离条件,考察该色谱分离条件下烃基黄原酸的分离情况,流动相为20%乙腈-80% 0.05 mol/L乙酸铵溶液(pH为9.5)。由于实验室设备仅配备了质谱检测器,没有配备紫外检测器,于是采用质谱检测方式,结果见图7。
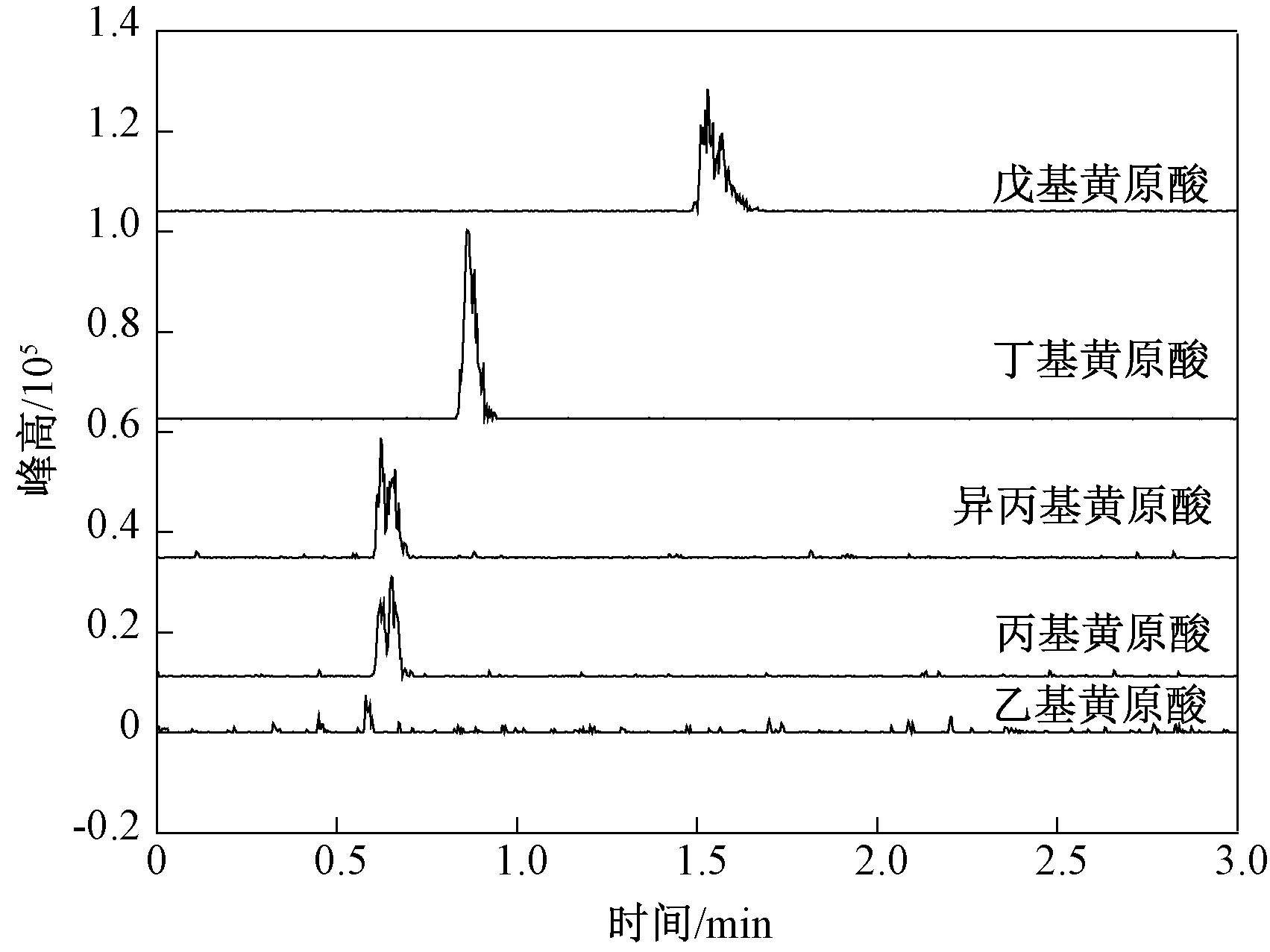
图7 烃基黄原酸总离子流Fig.7 TIC chromatograms of alkyl xanthic acid
同时,保持文献[22]其他色谱分离条件不变,将流动相中的乙酸铵溶液换成pH相同的氨水溶液,即流动相为20%乙腈-80%氨水溶液(pH为9.5)。并在此条件下采集几种烃基黄原酸的总离子流图,见图8。

图8 等度洗脱模式下的烃基黄原酸总离子流Fig.8 TIC chromatograms of alkyl xanthic acid achieved by isocratic elution program
将图7与图8对比,可知流动相中乙酸铵的加入抑制了烃基黄原酸的质谱信号且峰型不理想,但保留时间分布还是有较大区别,可见文献[22]的分析条件下,乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸并不干扰丁基黄原酸的测定。但从丙基黄原酸和异丙基黄原酸的保留时间分布区域来看,两者保留时间位于同一区域。而图8显示,氨水溶液(pH为9.5)∶乙腈=80∶20等度洗脱模式下,几种烃基黄原酸的保留时间都很短,且丙基黄原酸和异丙基黄原酸的保留时间相同,丁基黄原酸和戊基黄原酸的保留时间也相同。对比图8与图2可以发现,图2中工业品丁基黄原酸峰旁边出现的峰(保留时间为0.98 min)并没有在图8中出现。可见,氨水溶液(pH为9.5)∶乙腈=80∶20等度洗脱模式下,丁基黄原酸的紫外吸收检测会受到图2中保留时间(0.98 min)对应物质的干扰以及戊基黄原酸的干扰。
5.6 液相色谱质谱法
近年来,液相色谱质谱仪因其灵敏度高、选择性好等优点发展相当迅速,并且随着环境监测部门仪器装备水平的提高,在环境监测和分析领域应用广泛[24-26]。杨坪[27]率先将液相色谱-质谱应用于丁基黄原酸的测定,该法直接进样20 μL,丁基黄原酸在0.05~100 μg/L范围内线性良好,方法检出限为0.05 μg/L,加标浓度为0.05~100 μg/L时,相对标准偏差<4%,回收率为89%~107%。刘景泰等[28]利用超高效液相色谱质谱测定了地表水中丁基黄原酸。该法以氨水溶液(pH约为9.5)∶乙腈=80∶20等度洗脱,Waters ACQUITY UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7 μm)分离,三重四极杆串联质谱多级反应监测模式分析,当直接进样10 μL时,丁基黄原酸在0.5~50.0 μg/L浓度范围内线性良好,方法检出限为0.2 μg/L,空白样品中加标2.0 μg/L丁基黄原酸,回收率为90%以上。目前,《水质 丁基黄原酸的测定 液相色谱-三重四极杆串联质谱法》[9]已颁布实施。该方法中流动相A为乙腈,流动相B为氨水溶液(pH约为9.5),乙腈初始比例为20%,保持1.5 min,1 min内升高到90%,保持1 min,0.5 min内降为20%,保持2 min。笔者采用《水质 丁基黄原酸的测定 液相色谱-三重四极杆串联质谱法》[9]中的分离条件考察了几种烃基黄原酸的分离情况,结果见图9。
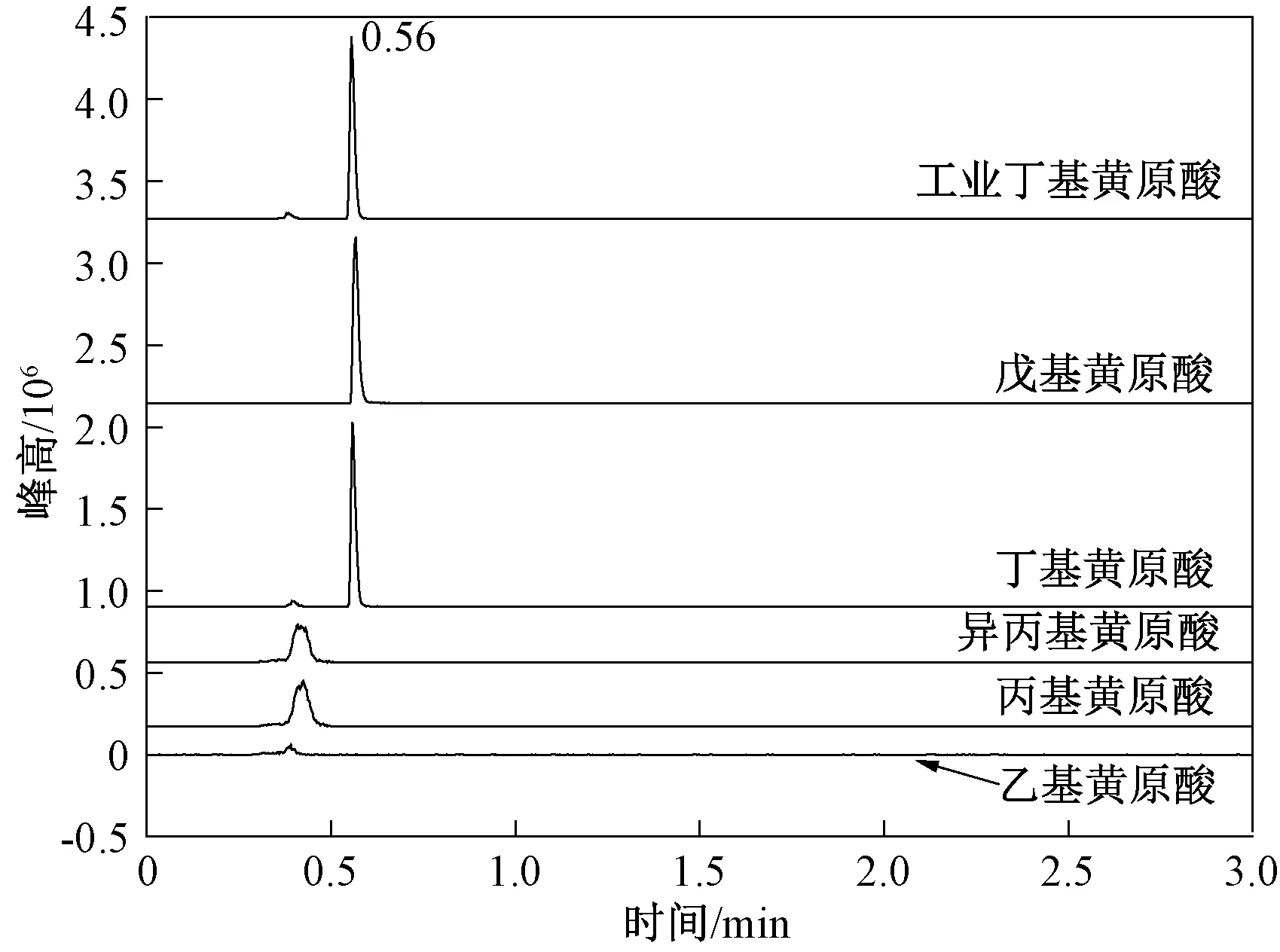
图9 梯度洗脱模式下的烃基黄原酸总离子流Fig.9 TIC chromatograms of alkyl xanthic acid achieved by gradient elution program
在此分离条件下,丁基黄原酸、戊基黄原酸色谱行为一致,保留时间均为0.56 min,出峰较快,事实上丁基黄原酸和戊基黄原酸色谱峰出完时,流动相还是20%A~80%B等度,即几种烃基黄原酸色谱分离时并没有发挥梯度洗脱的作用。笔者对该方法中的流动相梯度进行改进。在改进的流动相梯度下,目标物丁基黄原酸与乙基黄原酸、丙基黄原酸、异丙基黄原酸和戊基黄原酸得到了很好的分离。对比图9与图2中的丁基黄原酸总离子流图还可以发现,图2中工业品丁基黄原酸峰旁边出现的峰(保留时间0.98 min)并没有在图9中出现,由此看来,《水质 丁基黄原酸的测定 液相色谱-三重四极杆串联质谱法》[9]中的流动相分离条件也不能将图2中保留时间为0.98 min对应的物质与丁基黄原酸分离开。鉴于丁基黄原酸在反相固定相上保留弱,且易受到共存黄原酸干扰等问题,笔者推荐采用梯度方法进行液相色谱分离,初始流动相组成中有机相比例不宜过高,以增强丁基黄原酸在反相固定相上的保留。另外,液相色谱质谱分析中很容易受到样品基质的干扰,使得待测物信号受到抑制或增强。因此,复杂基体中丁基黄原酸的分析仍是液相色谱质谱分析法面临的难点。
6 总结及展望
正确区分丁基黄原酸及其盐,有效保存样品,采用低损失的前处理方法以及选择性好的分析仪器,有利于提高丁基黄原酸测定的准确性。建议《地表水环境质量标准》(GB 3838—2002)和《生活饮用水卫生标准》(GB 5749—2006)中明确丁基黄原酸以C4H9OCSSH计。水样采集后应调节pH至9~10,并尽快分析,最长不超过1 d。水样过滤首选PTFE材质滤膜。当水样检出丁基黄原酸时,铜试剂亚铜分光光度法、紫外分光光度法、气相色谱/气相色谱质谱间接测定法在选择性方面均不能满足丁基黄原酸准确测定的要求,需要借助液相色谱质谱法、离子色谱法以及液相色谱法等方法进一步确认。
通过对水质丁基黄原酸测定难点探讨,展望未来发展趋势如下:
1)高纯度丁基黄原酸标准品以及配套内标物质的研制。
2)标准文件以及标准分析方法本身需再完善,尤其是复杂基体情况下分析方法选择性和灵敏度的提升。
3)低损失的丁基黄原酸样品富集技术的研究,以满足水质丁基黄原酸监测对灵敏度的需要。
4)液相色谱质谱法、离子色谱法以及液相色谱法操作简便、选择性好、灵敏度高,在水质丁基黄原酸测定方面具有更好的应用前景。分光光度法、气相色谱/气相色谱质谱间接测定法与液相色谱质谱法、离子色谱法、液相色谱法配合使用将是提升水质丁基黄原酸监测准确性的重要途径之一。
致谢:感谢攀枝花市环境监测中心站陈美芳教授级高级工程师和四川省生态环境监测总站赵云芝高级工程师对研究工作的指导。
猜你喜欢
能源化工(2021年3期)2021-12-31 11:59:23
世界农药(2021年3期)2021-12-09 13:01:25
金属矿山(2021年8期)2021-09-09 10:30:42
化工环保(2021年3期)2021-06-17 08:06:48
世界有色金属(2018年9期)2018-07-12 10:59:50
化工生产与技术(2016年5期)2016-03-13 10:07:26
化学与生物工程(2015年8期)2015-12-28 05:42:30
中国塑料(2015年12期)2015-10-16 00:57:12
西南民族大学学报(自然科学版)(2014年2期)2014-03-16 05:08:36
化学与生物工程(2014年8期)2014-01-14 09:04:42
