
miR-155在炎症性肠病中的免疫作用机制研究进展
2019-09-13 09:23刘星星
世界华人消化杂志 2019年17期
朱 凤,范 恒,刘星星
朱凤,范恒,刘星星,华中科技大学同济医学院附属协和医院中西医结合科 湖北省武汉市 430022
核心提要: miR-155通过Jarid2/notch1信号通路及促进2型巨噬细胞分化来调节TH17分化, 并通过抑制细胞毒T淋巴细胞相关抗原4调节Treg、通过细胞信号传导的抑制因子1调节肠成纤维细胞和肌成纤维细胞以及通过调节DNA双链断裂沉积来影响炎症性肠病发生发展.
0 引言
炎症性肠病(inflammatory bowel disease, IBD)为累及回肠、结肠、直肠的一种特发性肠道炎症性疾病, 主要包括溃疡性结肠炎(ulcerative colitis, UC)和克罗恩病(crohn's disease, CD), 其病因及发病机制尚未完全明确,目前普遍认为IBD的发病机制是由遗传、环境因素和肠道菌群共同作用, 激活遗传易感个体肠道黏膜的免疫应答, 引发一系列的炎症反应. 除了明显的遗传危险因素, 全基因组关联研究已经确定了许多常见的基因调控靶点, 尽管在此方面研究激烈, 炎症基因表达的调控仍然没有完全清楚, 而了解这些基因是怎样监管、调控IBD的发生发展具有重要意义.
microRNA是广泛存在于真核生物中的非编码小RNA, 长度为19-24nt, 它能在转录后水平抑制靶基因的表达或翻译. 近年来研究显示, miR-155是一个典型的多功能基因, 由其下游基因介导, 参与多种生理病理过程, 如炎症、免疫和肿瘤的发生发展. 本文就miR-155对IBD的免疫调节作用的研究进展作一综述.
1 IBD
IBD是一种慢性非特异性肠道炎症性疾病, 包括UC和CD, 其临床表现以腹痛、腹泻及黏液脓血便为主, 症状易反复发作, 难以治愈[1]. 该病多发于青少年, 在欧美国家发病率较高. 但随着中国经济社会的发展及人民生活方式的改变, 我国发病率较前明显增加[2]. IBD的病因尚不明确, 多数学者认为, IBD是由多因素综合作用导致. 因此, 找到治疗IBD的有效的治疗方法亟待解决.目前, IBD的医疗管理的主要依据是5-氨基水杨酸制剂,皮质类固醇, 硫嘌呤, 甲氨蝶呤, 抗肿瘤坏死因子, 抗α4β7整联蛋白和抗白细胞介素(interleukin, IL)-12/IL-23疗法. 发现涉及IBD发病机制的新途径导致新药靶向Janus激酶/信号转导物和转录激活因子, IL-6, 鞘氨醇-1-磷酸和磷酸二酯酶4等. 这些新疗法可能会带来更有利的安全性[3]. 近来许多研究表明miR-155在IBD的发病机制中占重要地位, 而了解发病机制可以更好地选择治疗靶点.
2 miR-155的生物学作用
miR-155是一个典型的多功能miRNA, 越来越多的实验研究表明miR-155参与了炎症、免疫、肿瘤及血细胞生成等多种生物学过程. miR-155位于人类21号染色体的非编码转录本B细胞整合簇基因(B-cell integration cluster, BIC)第三个外显子内[4], 其表达水平受BIC的转录水平和miRNA加工等调控. BIC是一个不含开放读码框的基因, 过表达BIC可促进细胞异常增殖. Leng等[5]证实miR-155被编码在BIC, miR155HG的区域内, 该区域最初被鉴定为禽白血病病毒的常见整合位点. miR-155参与多种炎症病变过程. Lu等[6]研究发现miR-155在脂多糖(lipopolysaccharide, LPS)刺激的单核细胞中上调.Hu等[7]发现miR-155抑制剂可降低LPS诱导的巨噬细胞炎症和NF-κB通路活化, 但可增加细胞信号传导的抑制因子1(suppressor of cytokine signaling1, SOCS1)的表达, 并得出miR-155抑制剂可通过SOCS1/NF-κB通路减少巨噬细胞炎症, 减轻心肌梗死后内质网应激诱导的心肌细胞凋亡. Li[8]等研究表明miR-155-5p通过调节肿瘤蛋白p53诱导核蛋白1的表达来调节宫颈癌细胞的发育. 目前已有大量的实验证据表明miRNAs也可以由病毒基因组编码. 到目前为止, 已知的病毒miRNAs来源于双链DNA病毒—疱疹病毒属, 多瘤病毒属以及腺病毒属. 病毒miRNAs可以干扰受感染细胞的mRNAs, 从而调节基因表达. Wood等[9]发现Epstein-Barr(EB)病毒携带的两个基因(潜伏膜蛋白1和EB病毒核心抗原2)上调miR-155表达, 并且miR-155表达是EBV感染的B细胞生长所必需的. 我们显示EBV转录因子EB病毒核心抗原2通过激活miR-155宿主基因(miR-155HG)上游的增强子来上调miR-155表达, miR-155来源miR-155宿主基因. 而且研究显示EB病毒核心抗原2还通过增强子介导的干扰素调节因子4活化间接激活miR-155表达, 然后独立于EBNA2激活miR-155HG启动子和上游增强子. 综上可以看出, miR-155参与各种疾病的病理生理过程, 因此,对其在IBD中的作用有待深入研究.
3 miR-155通过Jarid2/notch1信号通路调节IBD
Th17/Treg失衡在IBD结肠黏膜免疫紊乱和炎症状态中起重要作用, 找到影响Th17细胞分化的机制将帮助我们找到治疗IBD的新靶点. Liu等[10]研究表明, miR-155在调节免疫系统功能中具有重要作用, 在IBD结肠组织中也被检测到显着上调. Escobar等[11]揭示miR-155的缺失导致Jarid2表达增加, 并增加核心蛋白复合体2(polycomb repressive complex 2, PRC2). PRC2招募, 减少了IL-22转录; 而在Jarid2缺陷型Th17细胞中, PRC2募集率降低.Mysliwiec等[12]报道Jarid2占据内源性Notch1基因座的调控区域, 表明Jarid2直接控制小鼠胚胎心脏组织中的Notch1表达, 由配体激活的Notch1释放出一个细胞内片段(notch1 intracellular domain, N1ICD)直接结合ROR-γt和IL-17启动子并调节Th17分化. Liu等[10]通过使用由慢病毒载体递送的miR-155抑制序列, 其显示抑制miR-155可以改善TNBS诱导的实验性结肠炎. 进行相关检测发现在TNBS+miR-155抑制剂组中, Th17细胞在脾脏和肠系膜淋巴结中的比例和结肠组织中Th17细胞相关细胞因子IL-6, IL-17A, IL-17F和IL-21的水平显着减少, 表明抑制miR-155调节Th17细胞的功能. 通过免疫组织化和蛋白印迹法发现Jarid2显著升高, miR-155抑制和notch1表达与Jarid2呈负相关. 这项研究表明通过调节抑制miR-155可以改善TNBS诱导的结肠炎, 且Th17细胞分化和功能与Jarid2/notch1密切相关. 因此我们可以知道,miR-155可以通过Jarid2/notch1调节TH17分化, 从而影响IBD发生发展.
4 miR-155通过调节M2分化来调节IBD
IBD与肠黏膜中的先天性和适应性免疫应答的失调有关. MicroRNA(miR-155)在许多免疫细胞中表达并起作用. 除了其在适应性免疫中的功能外, miR-155是巨噬细胞、树突状细胞甚至上皮细胞中先天免疫应答的关键调节剂. 由于内在分子调节和外在环境差异, 未分化的巨噬细胞可以极化为促炎性M1巨噬细胞或抗炎M2巨噬细胞, 巨噬细胞对维持肠道稳态至关重要[13]. 在结肠炎免疫反应的早期阶段, 血液单核细胞以细胞表面趋化因子受体2依赖性方式从中招募炎性巨噬细胞, 并在发炎的黏膜中积累和产生促炎介质[14]. 如果炎症性巨噬细胞反应不受控制, 则随后引起适应性免疫反应和炎症性T细胞, 包括Th1细胞和Th17细胞, 被招募致病现场, 这些细胞可进一步加重结肠炎症损伤. 在一些条件下, 肠道中的巨噬细胞可能被迫通过内在因素和外在因素转变为M2表型, 诱导的M2巨噬细胞在化学诱导的结肠炎中显示出巨大的治疗潜力[15,16]. Li等[17]发现miR-155在结肠炎中是一种强有力的调节因子去调节巨噬细胞极化(图1), 其缺乏可导致巨噬细胞M1型转变成M2型. M2在结肠中的优势可导致肠道免疫细胞增殖受抑制并抑制CD4+T细胞向Th1和Th17极化. 因此, miR-155可通过调节M2极化来调节TH17分化并影响IBD的发生发展.
5 miR-155通过抑制CTLA-4来调节Treg细胞,进一步调节肠道免疫及IBD发生发展
研究表明, miR-155的过表达增强了CD8+T细胞的抗原特异性免疫应答和克隆增殖, 它的缺陷导致T细胞的抗肿瘤免疫功能减弱, 并可通过细胞毒T淋巴细胞相关抗原4(cytotoxic T lymphocyte-associated antigen-4, CTLA-4)抗体重新储存[18,19]. CTLA-4是免疫检查点阻滞(immune checkpoint blocker, ICB)治疗的一个重要的目标, 这表明miR-155对CTLA-4具有潜在的调节作用. 最近, 研究表明miR-155以竞争内源性RNA(ceRNA)的形式与CTLA-4 mRNA 3'UTR结合, 增强辅助性T细胞的增殖反应[20].CTLA-4也被认为在IBD的进展中起关键作用. 例如,SOCS1缺陷小鼠中的CTLA-4下调会诱导严重的IBD[21],并且CTLA-4缺乏与早发型CD之间存在相关性[22], 此外, CTLA-4-ICB治疗可引起严重胃肠道溃疡的不良反应[23]. CTLA-4是一种由T细胞表达的共抑制分子, 是一种重要的免疫调节因子和炎症抑制剂[24]. Chao等[25]通过研究表明, miR-155模拟剂和抑制剂可分别下调和上调CTLA-4蛋白和mRNA的表达. 这些结果表明miR-155可直接靶向抑制CTLA-4的表达. 滤泡调节性T细胞(follicular regulatory T cells, Tfr)通过调节滤泡辅助性T细胞(follicular helper CD4 T cells, Tfh)依赖性生发中心反应, 抑制B细胞反应并防止幽门自身免疫介导的肠损伤.Tfr细胞的缺乏可能是CTLA-4缺乏引起的IBD免疫抑制的原因. miR-155可以靶向CTLA-4在cTreg和Tfr中的表达, 直接抑制Tfr细胞的产生并促进增强生发中心B细胞活化和自身抗体过量产生. 因此可以说明, miR-155可以通过调节CTLA-4表达来调节肠道免疫功能及肠黏膜损伤, 为治疗IBD提供新的思路.
6 miR-155作用于SOCS1来调节IMF产生,影响IBD
越来越多的证据表明间充质细胞, 如肠成纤维细胞和肌成纤维细胞(intestinal myelofibrosis, IMF), 正在积极参与肠黏膜的炎症过程[26]. 持续炎症期间其表型和功能的稳定改变可通过促进组织破坏、支持免疫细胞的募集,并通过产生各种细胞因子保留和活化免疫细胞等方式向慢性肠道炎症转变. IMF是感应和应对各种压力来保持肠黏膜稳态的塑料细胞[27]. 然而, 在IBD中, IMF在持续炎症性刺激时获得活化的表型并大量增殖, 导致不必要的细胞外基质重塑, 并产生一个过量的可溶性介质, 如炎性细胞因子, TGF-β1和Wnts配体, 其深刻影响邻近的上皮细胞、间充质和免疫细胞. Pathak等[28]研究揭示, 分离正常对照组、UC组、CD组结肠的IMF, 发现与对照组及CD组患者相比, miR-155在UC患者的IMF中显著上调. 它在IMF中的表达受促炎调节信号的作用,如TNF-α和LPS, 但不是促纤维蛋白原介质, 如TGF-β1.miR-155在对照IMF中的过表达表明了UC衍生的IMF的促炎表型, 而在UC衍生的IMF中miR-155敲低可以纠正它们的促炎表型. 而且, 进一步研究发现了miR-155直接靶向SOCS1, 其表达显着下调UC衍生的IMF. IMF是导致IBD黏膜损伤的关键细胞群, miR-155在对照IMF中增强细胞因子的异位表达和释放, 而它下调SOCS1表达, UC-IMF中的miR-155敲低减少细胞因子的产生并增强SOCS1表达, 荧光素酶基因测定报告证明miR-155直接靶向SOCS1. 而且, 沉默控制IMF中SOCS1的表达显着增加IL-6和IL-8的释放. 总之, miR-155可以通过抑制SOCS1表达, 调节IMF炎症表型, 从而影响UC发生发展.
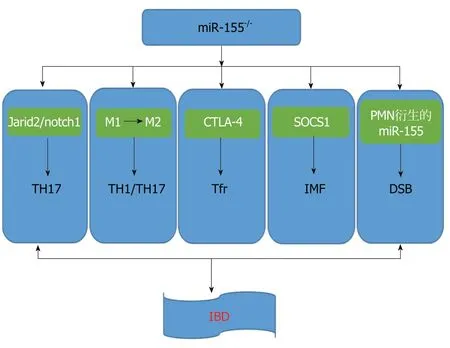
图1 miR-155在IBD中的免疫作用机制.
7 PMN衍生的miR-155调节DSB积累影响肠道炎症反应,调节IBD进程
由于炎症加剧导致的上皮损伤反应是胃肠道的常见病理特征, 包括IBD[29,30]. 肠黏膜中的炎症反应不可避免地导致中性粒细胞的募集[多形核白细胞(polymorphonuclear leukocyte, PMN)]. 黏膜上皮细胞炎症部位的PMN的募集, 其对组织微环境和促进上皮恢复的意义重大[31]. PMN在宿主防御中起着至关重要的作用, 失调的PMN募集可导致组织损伤. 因此, 在IBD中,肠黏膜中的PMN的数量与疾病严重程度相关[32]. PMN的病理学影响主要归因于释放可溶性介质, 包括基质金属蛋白酶(matrix metalloproteinase, MMPs), 中性粒细胞弹性蛋白酶和髓过氧化物酶[33]. 虽然氧化还原信号是细胞更新、迁移和增殖的重要组成部分[34], 但过量的活性氧(reactive oxygen species, ROS)可以破坏组织平衡[35].ROS对DNA糖磷酸盐骨的攻击可以诱导单链和/或双链断裂(DNA double-strand breaks, DSBs)的形成[36]. DSB积累可诱导细胞凋亡或衰老, 导致基因组不稳定, 是癌症发生的标志[37]. 高效的上皮伤口愈合对于组织稳态修复至关重要[38]. 最近研究表明, 免疫细胞, 特别是PMN, 有助于调节涉及上皮愈合反应的关键过程, 包括迁移和增殖[39]. 这种调节的新机制, 即组织浸润PMN释放细胞外囊泡或微粒(PMN-MPs), 提供各种生物功能分子, 如MMPs或过氧化物酶可主动调节上皮屏障功能和伤口愈合[40,41]. Butin-Israeli等[42]使用IBD临床样本和在体外和体内损伤模型中, 显示PMN衍生的miR-23a和miR-155通过诱导核纤层蛋白B1依赖性复制叉崩溃和抑制同源重组(homologousrecombination, HR), 针对HR调节器重组蛋白A, 促进DSB的积累. 受损上皮中的DSB积累导致结肠愈合和基因组不稳定性受损. 在培养的肠上皮细胞和急性损伤的黏膜中, 靶向抑制miR-23a和miR-155减少了PMN的有害作用并增强组织愈合反应. 所以, 抑制PMN衍生的miR-155可以减少DSB积累, 促进肠黏膜愈合, 可以作为治疗IBD的新思路.
8 结论与展望
肠黏膜屏障破坏、通透性增加、黏膜NF-kB活化、炎性细胞因子分泌增多等均是IBD发病及反复发作、迁延不愈以致癌变的原因. 由上可知, 抑制miR-155可以促进Jarid2/notch1信号通路传导, 从而抑制TH17产生, 调节肠道免疫功能; miR-155-/-可以促进M1向M2转化, 而M2可抑制免疫细胞增殖, 减少TH1、TH17产生; 同时,miR-155还可以抑制CTLA-4及SOCS1, 调节IMF炎症表型, 从而影响IBD发生发展; PMN衍生的miR-155可以促进DSB在肠道黏膜沉积, 加重黏膜组织损伤, 从而加重IBD症状(图1). miR-155可以通过各种机制达到影响IBD的作用, 但它们不是独立的, 而是共同发挥作用, 但目前起主要作用的机制并未研究透彻. 由此, 我们可以知道miR-155是一个重要的、多效的microRNA. 尽管microRNA许多的功能和表达调节机制至今没有完全清楚, 但是miR-155在许多炎症及肿瘤组织的过表达让我们意识到其在炎症和肿瘤诊断及治疗中的重要意义.Borghei等[43]使用单链DNA探针和DNA/RNA异源双链相互作用的碲化镉量子点聚集的miRNA识别的光谱方法用于测定人乳腺癌MCF-7细胞和正常人胚肾细胞系中的miR-155. 因此, miR-155在IBD的发生发展中占据重要地位, 为后期治疗IBD及相关肿瘤疾病上拓宽了视野, 也提供了新的治疗思路.
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
中老年保健(2021年8期)2021-12-02
中老年保健(2021年8期)2021-08-24
天津医科大学学报(2021年4期)2021-08-21
汽车维修与保养(2021年8期)2021-02-16
中国民间疗法(2020年22期)2021-01-07
医学新知(2019年4期)2020-01-02
医学新知(2019年4期)2020-01-02
中国内镜杂志(2017年2期)2017-03-20
中国卫生标准管理(2015年3期)2015-01-27
