
锂电池研究中的拉曼/红外实验测量和分析方法
2019-09-11 03:44:50孙姝纬赵慧玲郁彩艳
储能科学与技术 2019年5期
孙姝纬,赵慧玲,郁彩艳,白 莹
(河南大学物理与电子学院,河南 开封 475004)
拉曼光谱(Raman spectroscopy,简称Raman或RS)与红外光谱(infrared spectroscopy,简称IR)是两种常见的物理性质表征方法,在物理、化学、材料、能源、考古、天文、地质等领域有着非常广泛的应用。随着锂电池的广泛应用,在该领域的科学研究和工程化中也发挥了越来越重要的作用,以下分别简述两种光谱技术的发展历程及应用范围。
1928年印度物理学家拉曼在研究液体光散射时,发现散射光中除有与入射光频率相同的瑞利散射线外,在瑞利散射线ν0的两侧还有红伴线和紫伴线,且伴线与瑞利线的频率差值与入射光的频率无关[1]。这些特征使得拉曼光谱被很快应用于研究介质的分子结构,也导致了拉曼光谱学的迅速兴起及蓬勃发展。
然而,由于拉曼散射光强极弱,仅相当于入射光强度的百万分之一左右,极大地制约了拉曼光谱技术的应用和发展。直到近半个世纪由于激光器、全息光栅、光电倍增管、CCD探测器、计算机和数字计算技术的诞生和迅猛发展,才在20世纪90年代诞生了商业化的显微拉曼光谱仪,使拉曼光谱测量很快成为一种在操作难度上与发射光谱和吸收光谱相当的常规光谱技术,被应用于不同成分(组分)物质的鉴别、结构的测定(如晶体结构)和相变研究等。另外,表面增强拉曼光谱技术、拉曼光谱成像技术也为拉曼光谱的应用开辟了更广阔的空间。
同样在20世纪初,研究者们首次获得了材料的红外光谱并对红外吸收的特征频率进行了研究。之后红外光谱得到了广泛应用,并建立了红外光谱特征频率库。但在20世纪60年代中后期,随着各种新分析技术的出现,加之经典近红外光谱分析技术暴露出的灵敏度低、抗干扰性差的弱点,人们逐渐淡漠了该技术在分析测试中的应用。此后,近红外光谱再次进入了沉默期。
20世纪70年代,美、德等国研制并推出第三代红外分光光度计,它没有分光元件,而是通过干涉仪与计算机紧密联系组成的新型傅里叶变换红外光谱仪,这一发现促进了近红外光谱技术的推广。到20世纪80年代后期,随着计算机技术的迅速发展,带动了分析仪器的数字化和化学计量学的发展,通过化学计量学方法在解决光谱信息提取和背景干扰消除方面取得了的良好效果,加之近红外光谱在测样技术上的独特优势,使人们重新熟悉了近红外光谱的价值,近红外光谱在各领域中的应用研究陆续展开。进入20世纪90年代,近红外光谱在产业领域中的应用全面拓展,有关近红外光谱的研究及应用文献几乎呈指数增长,成为发展最快、最引人注目的一门独立的分析技术。
红外吸收峰的位置反映了分子结构上的特点,可以用来鉴别未知物的结构组成或确定其化学基团,吸收谱带的强度与化学基团的含量有关,可用于进行定量分析和纯度鉴定。另外,在化学反应的机理研究中,红外光谱也发挥了一定的作用。但其应用最广的还是未知化合物的结构鉴定:红外光谱不但可以用来研究分子的结构和化学键,如力常数和分子对称性,而且还可以作为表征和鉴别化学物种的方法。
本文在分别介绍拉曼光谱和傅里叶变换红外光谱原理的基础上,重点讨论两种光谱技术在锂电池研究中的应用,包括测试方法、测试仪器及参数、测试流程、测试注意事项等,并结合实际研究案例,具体分析拉曼光谱/傅里叶变换红外光谱以及上述两种手段与一些测试方法联用技术在锂电池中的应用。
1 实验原理
拉曼光谱和傅里叶变换红外光谱利用的是分子振动光谱的不同特性。前者主要对应于非极性基团与骨架的对称振动,后者则主要反映了极性基团的非对称性振动,两种方法相互补充,可以用来研究锂电池电极材料或界面在充放电过程中的组分和结构的变化。
1.1 拉曼光谱
拉曼光谱法是利用激光束照射样品时发生散射现象,而产生与入射光频率不同的散射光谱所进行的分析方法。它属于分子振动光谱,但它得到的振动能级信息不是来自样品对光的吸收,而是来自样品的散射。入射光可以看成是具有特定能量的光子,当与样品分子相碰撞时,若产生的散射光频率与入射光频率相同,则能量保持不变,这种弹性散射称为瑞利散射;若产生的散射光频率与入射光频率不同,则为非弹性碰撞,这种散射称为拉曼散射。其中,若散射光频率低于入射光,即入射光将一部分能量传递给样品分子,发生斯托克斯散射;若散射光频率高于入射光,即入射光从样品分子中获得一部分能量,发生反斯托克斯散射。常温下,斯托克斯散射的强度比反斯托克斯散射高得多。因此,在拉曼分析中,通常使用斯托克斯散射线。斯托克斯光的频率与入射光的频率之差称为拉曼位移(Raman shift)[2]。
拉曼光谱图通常是以拉曼位移为横坐标,谱带强度为纵坐标表示散射强度。拉曼位移就是分子的振动或转动频率。不同化合物的分子具有不同的拉曼位移、拉曼谱线数目和拉曼相对强度,这是对分子基团定性鉴别和分子结构分析的依据,因此可以通过晶格振动的特征峰及峰宽来判断晶体结构及其对称性。而对于同一个化合物,拉曼散射强度与其浓度呈线性关系,可以据此做一些定量或者半定量的测量。拉曼光谱出现在可见光区,其拉曼位移一般为40~4000 cm-1。
拉曼光谱适用于对称结构极性较小的分子[3],例如对于全对称振动模式的分子,在激发光子的作用下,会发生分子极化,产生拉曼活性,且活性很强。通常准备测试的样品无需经过特殊处理,所以在锂电池体系的测量中有着广泛的应用。对于拉曼光谱灵敏度不够的情况,可以使用一些Au或Ag等活性衬底在样品表面进行处理,由于在这些特殊金属纳米颗粒的表面电磁场的增强导致吸附分子的拉曼光谱信号增强,称之为表面增强拉曼散射(surface enhanced raman scattering,SERS)。
1.2 傅里叶变换红外光谱
红外光谱使用的波段与拉曼光谱类似,不少拉曼活性较弱的分子也可以使用红外光谱进行表征。红外光谱属于分子吸收光谱,可作为拉曼光谱的补充。当样品受到频率连续变化的红外光照射时,分子吸收其中特定的与分子振动能级差对应频率的辐射,故红外光谱又称为分子振动光谱。由分子振动引起偶极矩的变化,使振动能级从基态跃迁至激发态,在对应于该特征频率的位置出现吸收峰,记录下来的即是该分子的红外光谱图。红外光谱通常是以吸收光的波长或波数(wavenumber,cm-1)为横坐标,以透过百分率为纵坐标表示吸收强度,峰强与分子跃迁的概率或分子偶极矩有关。一般来说,极性较强的分子或基团对应的吸收峰也较强;分子的对称性越低,所产生的吸收峰越强[3]。
红外光谱主要用于化合物鉴定与分子结构表征,也可用于定量分析。与拉曼光谱相比,极性官能团的红外光谱更强,如C=O伸缩振动的红外光谱比相应的拉曼光谱更为显著。根据红外光区波长的不同,可以将红外光谱划分为三个区域:① 近红外区(即泛频区):波数在4000 cm-1以上的区域,主要测量O—H、C—H、N—H键的倍频吸收;② 中红外区(特征振动区/指纹区):波数范围在400~4000 cm-1,也是研究和应用最多的区域,主要振动频率是基频和指纹频率;③ 远红外区:波数在400 cm-1以下的区域,测量的主要是气体分子的转动信息。与拉曼光谱不同的是,由于水是极性很强的分子,它的红外吸收非常强烈,因此水溶液不能直接测量红外光谱,而且普通红外光谱测试前通常需要在干燥环境下将样品与KBr研磨压片。
一般情况下,红外光谱数据需要进行傅里叶变换处理,因此红外光谱仪和傅里叶变换处理器通常联合使用,得到的光谱称为傅里叶变换红外光谱(FITR)。在锂电池电解液的研究中,使用红外光谱手段的报道较多。
拉曼光谱与红外光谱均能获得有关分子内部各种简正振动频率及有关振动能级的情况,从而可以用来作官能团鉴定,但两者产生的原理和机制都不相同。在分子结构分析中,拉曼光谱与红外光谱相互补充,一些红外光谱无法检测的信息在拉曼光谱中能很好地表现出来。红外光谱侧重于检测基团,适用于极性键,多用有机物测量;拉曼光谱侧重于检测分子骨架,适用于非极性键,有机无机均可测试。拉曼光谱与红外光谱的对比如表1所示。
1.3 联用技术
拉曼光谱和红外光谱各有优势,但同时也存在一定的局限性,在实际应用和分析中以这两种表征为基础,拓展出新的表征手段和测试技术势在必行,于是出现了两种或两种以上测试仪器联用的方法。常见的联用手段有:原子力显微镜(atomic-force microscopy,AFM)与拉曼联用(AFM-Raman)、光致发光(photoluminescence,PL)与拉曼联用(PL-Raman)、扫描电子显微镜(scanning electron microscope,SEM)与拉曼联用(SEM-Raman)、热重(thermogravimetry,TG)与傅里叶变换红外光谱联用(TG-FTIR)、气相色谱(gas chromatography,GC)与傅里叶变红外光谱联用(GC-FTIR)、质谱(mass spectrum,MS)与傅里叶变换红外光谱联用(MS-FTIR)等。
2 实验测试方法
2.1 常规测试
2.1.1 电极材料测试
锂电池电极活性材料通常为固体粉末状样品,因而在Raman测试时,将少量样品放在样品台上直接进行测试即可。而由于水的极性很强,FTIR测试极易受到水分子的影响。因此在FTIR测试前,需要将待测样品充分干燥,然后取1~2 mg样品在玛瑙研钵中研磨成细粉末并与提前充分干燥的溴化钾粉末(用作红外光学窗口,AR级,约100 mg,粒度200目)混合均匀,装入模具内,在压片机上压制成片进行测试,整个制备过程须在干燥环境下完成。
2.1.2 电解质测试
目前绝大多数商用的锂电池电解质是液体。液体电解质的Raman测试仅需吸入毛细管或者滴加在载玻片上(也可以覆盖一层超薄盖玻片),然后准确聚焦,直接在其本征状态下测试即可;对于凝胶电解质,可直接滴加在载玻片上测试即可。而对于FTIR测试来说,依然需要在无水环境下制备样品后再行测试。常见的制备方法是液膜法,具体操作是:对于油状或黏稠液体(如凝胶),直接涂于KBr晶片上测试;流动性大且沸点低的液体(≤100 ℃),可夹在两块KBr晶片之间或直接注入厚度适当的液体池内测试;而对于水溶液样品,可用有机溶剂萃取水中的有机物,待溶剂挥发后,所留下为液体的遵循上述办法,为固体的则用KBr压片法测试。锂电池固体电解质近些年发展迅速,对于固体电解质的样品测试,参照电极材料的测试方法。
2.2 特殊方法测试
2.2.1 充放电循环后电极材料的测试
为研究充放电过程对电极材料表面组分和结构的影响,可以将循环后的电极材料经一定处理后进行光谱测试。目前使用的模拟电池在进行光谱测量前,需在装配好一定的电极-电解液体系后,对电池进行充放电模式以及充放电状态的设置;然后,将目标状态下的电池在手套箱中卸开,取出电极材料,用溶剂反复浸泡清洗,真空干燥后进行测试。如果倾向于对活性电极表面副反应产物组分的表征,可以在最初涂制电极片的过程中仅用活性材料和溶剂制片,排除导电添加剂和黏结剂本身以及它们可能参与的副反应的影响,且在Raman测试前,在手套箱中将清洗干燥后的极片用真空封脂或密封胶带密封在超薄石英片和载玻片之间,再进行测试。普通极片的Raman测试时仅将电极片置于拉曼光谱仪样品台上进行测试即可。而所有的FTIR表征均需将电极片上的活性材料与干燥的溴化钾在玛瑙研钵中混合均匀,再压片进行测试,这里的整个过程依然需要在干燥环境下完成。若是对电极表面副反应产物的FTIR测试,应尽量减少样品暴露在空气中的时间。
2.2.2 原位拉曼/红外测试
原位研究是指用相应的分析或检测仪器对正在反应变化的实验体系进行直接测量,而无需改变整个体系的实际工作状态,从而可以获得真实准确的结果。原位拉曼/红外电池的具体制作方案:负极内装有柔性导电柱,将其与底座连接,正极与上盖相连,并在上盖的顶端设置一个窗口,此窗口为透光的石英片,通过衬套压紧与上盖密封连接,此衬套与绝缘套两者可拆卸连接,而绝缘套又与底座之间可拆卸密封连接。另外,对于空气敏感的样品,也可以使用此装置进行测试。原位拉曼/红外电池的示意图及光学照片如图1所示。
2.2.3 表面增强测试
金属纳米颗粒具有特殊的光学性质,即当入射光线照射到有一定粗糙度的贵金属表面时,在金属表面吸附的物质的拉曼及红外信号均可得到增强,这种现象称作表面增强拉曼散射(surface enhanced Raman spectroscopy,SERS)和表面增强红外吸收(surface enhanced infrared absorption spectroscopy,SEIRAS)。目前已有很多金属被证实可以用于制备活性衬底,其中Ag获得了最广泛的应用。常见的制备银衬底的方法有两种:液相还原法制备和固相法制备。液相还原法制备银衬底虽简单,却耗时较长,且同时需剧烈搅拌。相对来说,固相法制备方法如激光刻蚀,得到的衬底分布均匀、重复性好,但纳米银的粒径较大。
2.2.4 联用(couple)测试技术
将原子力显微镜(AFM)分析与拉曼集成在同一台显微镜上,组成AFM-Raman联用系统。原子力显微镜是利用针尖原子与样品表面原子间的范德华力来反馈样品表面形貌信息或探测样品微观力学特性。AFM-Raman联用系统可以在纳米尺度上提供样品的拓扑信息,并将这些信息与拉曼光谱成像提供的化学信息相结合,从而使得表征结果更完善。由于同属显微镜一族,在测试时直接对待测样品进行测试,无需制样。
热重(thermogravimetry,TG)与傅里叶红外光谱联用(TG-FTIR)技术能够实时并快速地测定样品在受热过程中发生的一些物理化学变化,还可以测定在热失重过程中生成的分解产物其化学成分。具体联用方式是:将热重分析仪的气体出口与FTIR的气体池联通,一般以高纯氩气做载气,并使传输管(transfer line)和红外气体池的温度分别维持在200 ℃和225 ℃,其他测试条件与单独测试相同。
气相色谱(gas chromatography,GC)一般和质谱(mass spectrum,MS)耦合后与傅里叶红外变换光谱一起组成联用(GC/MS-FTIR)。这样联用提供了一种原位表征锂电池气体释放的方法。具体做法如下:在充电过程中从电池收集逸出的气体,经由色谱仪的分析柱,根据极性和挥发性被分离,这样可以得到气体的组分。再连接到质谱仪,这时气体被电离,可以进一步证实组分。最后进入到与FTIR连接的光管,此光管内壁镀金,且管的两端以KBr窗片封口,实时监控红外吸收信号。最后将在色谱峰出现的时间段内存储的数据,以红外谱-气相色谱-功能团吸收强度三维图谱显示出来。
综上所述,对于各类材料在不同物理状态下的样品制备方法详见表2。
2.3 测试设备
用于拉曼或红外测试的仪器品牌和型号较多,如用于Raman测试的Renishaw、HORIBA等;用于FTIR测试的Thermo Fisher、Bruker Optics、Perkin Elmer等。常见的用于Raman的测试分析软件有LabSpec 6、wiRE2.0等;用于FTIR的测试分析软件有OMNIC、OPUS等。典型的用于Raman和FTIR测试的仪器规格参数分别如表3、表4所示。
由表3、4可知,不同的设备在分辨率、光谱范围、信噪比、积分时间/扫描速度等参数存在差别。其中仪器的各项分辨率需要重点关注,如时间/空间分辨率、光谱分辨率等。以拉曼光谱仪为例,时间分辨率指仪器在时间序列上能分辨的能力,它取决于探测器反应速度、激光器脉冲频率以及一起本身的灵敏度;空间分辨率指仪器能识别最小测试点的能力,它取决于激发光光斑大小和设备共焦性能(常规拉曼设备是通过扩束器技术、共焦技术等实现接近理论尺寸的小激光光斑,得到高空间分辨率性能和抑制杂散信号效果);光谱分辨率指仪器能分辨非常近似的结构的能力,这取决于设备中光栅的分辨率。而对于红外光谱仪的分辨率,与上述含义相同。由于锂电池中不同部分其样品组分、分子结构、官能团等均存在较大差别,实际测量时对各样品所处状态、制样标准等也有不同要求,因此,具体测试中需要结合理论知识和实际情况加以考量。

表2 不同物理状态下各类材料的样品制备方法Table 2 Preparation methods for different materials with various physical states
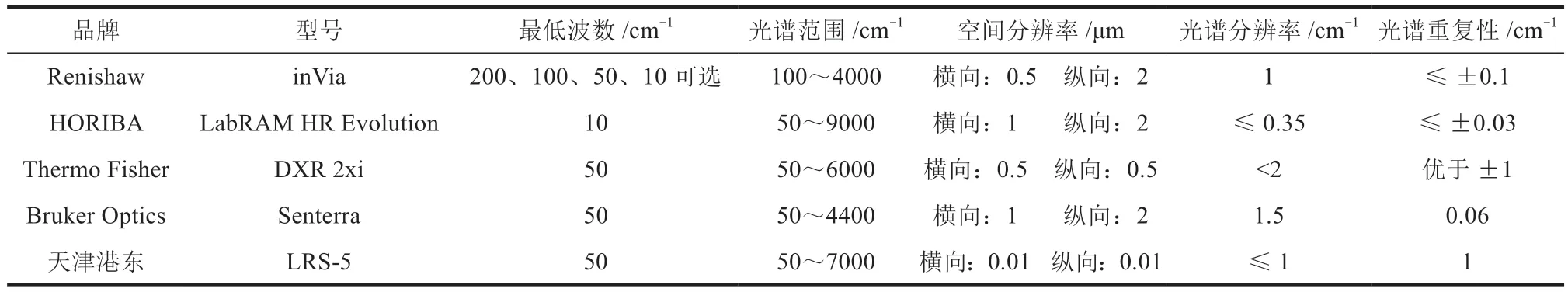
表3 拉曼测试光谱仪及其参数比较Table 3 Listed Raman spectroscopies and the corresponding parameters

表4 红外测试光谱仪及其参数比较Table 4 Listed FTIR spectroscopies and the corresponding parameters
2.4 测试流程
2.4.1 Raman测试与谱图处理流程
以LabRAM HR Evolution光谱仪器为例,介绍电极材料固体粉末样品的测试工步,如图2所示。
工步1:打开激光器进行预热,启动配套LabSpec 6软件,待CCD制冷(cool to operating temperature)温度稳定后,使用硅片校准。将样品放置在显微镜平台上,并使用可视装置(显微目镜或视频摄像头)正确调整样品位置;点击video图标,在Instrument setup的Objective选择合适物镜,将样品聚焦清楚。
工步2:点击stop all图标,停止白光图像采集;在Acquisition parameters下的Spectro中输入光栅位置,Range中输入采谱范围,Acq.time(s)中输入单次采谱曝光时间,Accumulation中输入循环次数,RTD time(s)中输入实时采集曝光时间。设置好参数后,点击光谱采集按钮,进行光谱采集。
工步3:光谱采集完毕后,单击save图标保存,一般将测试数据保存为.txt格式;也可以激活video窗口,保存为.16v格式的显微图像。如测试完毕,待CCD温度回到室温左右,依次关闭LabSpec 6软件,激光器及电源开关。
注意事项:①测试过程中的随机尖峰(或称宇宙射线)可以通过使用Spike Filter功能来自动去除;②采集过程中可通过去噪功能Denoise来降低光谱信噪比。
Raman光谱处理有很多种方法,常见的有扣背底、标峰位及峰位拟合等,下文以LabSpec 6软件为例,介绍上述三种处理方法,如图3所示。
方法1:扣背底。打开未扣背底的光谱,点击右侧工具栏Processing→Baseline correction,选择合适参数,在Baseline correction中点击Fit进行拟合(可尝试使用不同的参数拟合,多次重复直到背底线合适为止),点击Sub扣除背底。

图2 HR evolution拉曼光谱仪光谱采集基本流程Fig.2 Raman data collection by HR evolution

图3 Raman光谱处理基本流程Fig.3 Raman data processing by LabSpec 6
方法2:标峰位。打开光谱,点击右侧工具栏Analysis→Peaks,点击Find。若获得正常结果,则完成标峰位;若除峰位显示之外,还有其他一些峰或线存在,则需要在Analysis→Peaks→Display options下取消勾选Peaks、Sum和Res选框。
方法3:峰位拟合。若一些峰是由两个或两个以上的峰叠加而成,此时,如要获得精确的峰信息,就需要对峰位拟合。先截取需要拟合的峰,在可能是峰的位置点击标峰位,并在Shape下拉菜单中选择合适的峰形函数。在Fit options中设置拟合选项,点击Peaks中的Fit进行拟合(可多次点击Fit直至拟合总谱非常贴近实测曲线)。拟合完成后,在Analysis→Peaks中的Peak table获得各个峰的详细信息,其中p、a、w和ar分别代表峰位、峰强、半高宽和峰面积。最后点击copy peaks,将拟合数据粘贴到Excle里保存。
注意事项:上述光谱处理均以软件自动处理为例,此外还可手动进行数据处理。
2.4.2 材料的FTIR测试与谱图处理流程
常见的单独使用FTIR的测试光谱一般只用于定性分析,故较Raman测试更简单,这里以IRTracer-100为例,介绍电极材料固体粉末样品测试工步,如图4所示。

图4 IRTracer-100红外光谱仪光谱采集基本流程Fig.4 FTIR data measurement by IRTracer-100
工步1:开启光谱仪的电源预热,并查看样品仓内是否有遗留样品。启动Labsolutions IR软件,选择光谱扫描,点击功能条“仪器”中“初始化”,成功后进行参数设置。
工步2:点击“背景扫描”(扫描时样品架不能放有样品,有时需放置空白样品;如果做压片,则需放置纯KBr压片)。完成后,将制备好的压片放入样品室,点击“样品扫描”进行测试。

图5 FTIR谱图处理基本流程Fig.5 FTIR data processing by Labsolutions IR
工步3:样品扫描完毕后,图谱会自动保存到默认文件夹。若需打印图谱,点击“打印编辑”,选择需要打印的模板,点击“文件”→“打印”。测试完毕后,取出样品,依次关闭操作软件和电脑。
注意事项:①环境中的水蒸汽对红外图谱的结果有干扰,因此整个实验过程中应保持实验室干燥(环境湿度在30%~60%),不得带入有过多水源的样品;②实验室内应尽量少讲话或靠近样品仓呼吸,以保持二氧化碳浓度恒定,门窗尽量保持在闭合状态。
样品扫描完毕后可直接进行谱图处理,也可打开之前测试图谱进行处理,这里以Labsolutions IR软件为例,介绍基本谱图处理方法,如图5所示。
工步1:启动软件,打开任意谱图,点击图像上的X轴/Y轴,输入数字可变更范围;拖曳方框到需放大位置,即可放大部分谱线,工作区上部图谱中阴影部分表示放大部分在全谱中的位置;右键选择“全视图”即可还原;选择“图表”中的“Y轴设置”,以进行透过和吸光的转换。
工步2:点击 “处理”,可进行相关操作。如“峰值检测”:选择 “峰检测”,可根据峰强设置噪音(N)、阈值(T),点击“计算”,各峰波数显示在谱线下方;通过 “添加”或者“删除”进行增减,若对计算结果满意,点击确定,峰表即可弹出。
工步3:点击 “检索”,出现设置界面,常规:添加谱库;光谱:默认设置;峰:添加或删除峰;最后根据需要进行谱图检索类型。
3 数据及案例分析
通过拉曼及红外测试得到的光谱,可以直接获得分子结构(主要是分子官能团)的信息。拉曼光谱是一种散射光谱,能提供材料中振动、转动以及低频激发的非弹性散射的信息,而红外光谱是红外光子与分子振动、转动的量子化能级共振产生吸收而产生的特征光谱曲线。这两种方法均是从物质分子角度研究分子价键、官能团振动和转动能级跃迁状态的重要方法,相关谱学数据可用于确定分子结构和计算化学键键能、键长、键角等,还可用于研究物相结构变化、稳定性、表面现象和反应机理。合理的使用或配合使用两种测试方法可以帮助研究人员更好地理解材料在化学和/或电化学环境下的反应及变化,提升电池研发水平。以下结合具体案例介绍Raman/FTIR在锂电池中的应用。
测试及分析对象包括正、负极电极材料、电解质和黏结剂、固体电解质界面膜(solidelectrolyte interphase layer,简称SEI膜),着重讨论原位测试观测界面间的电化学过程,检测表面吸附物种、副反应中间产物和最终产物等。
3.1 常规表征
锂电池主要由电极材料、电解质、黏结剂等部分组成,许多研究者报道了采用拉曼和红外技术对电极材料、电解质、电极-电解质界面和黏结剂等在电化学环境下的组分和结构表征,下文扼要介绍这些部分的常规表征实例。
3.1.1 电极材料表征
电极材料是电池的核心,正负极活性物质是产生电能的源泉,是决定电池基本特性的重要组成部分。而电极材料组分或晶体结构的定性表征一般使用拉曼和红外的表征手段,常见的正极材料如以及近几年普遍关注的两种高电压材料:LiNi0.5Mn1.5O4[12-13]和富锂层状材料[14-15]。
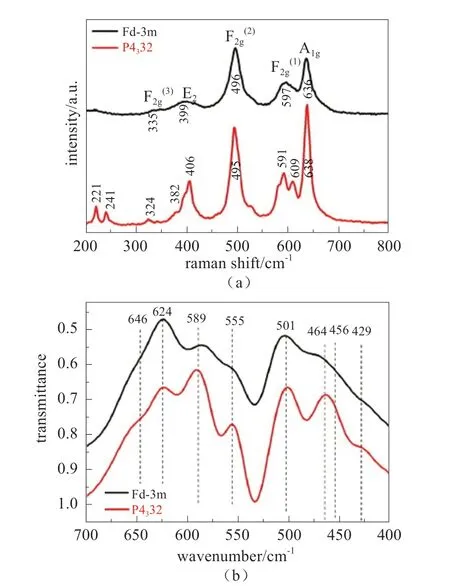
图6 (a)两种多晶型样品的拉曼光谱;(b)两种多晶型样品的FTIR光谱[13]Fig.6 (a) Raman spectra of two polymorphs; (b) FTIR spectra of two polymorphs[13]
拉曼和红外表征可区分同一样品的不同晶体结构,中国科学院物理研究所李泓等[13]使用不同合成温度,获得两种相结构LiNi0.5Mn1.5O4粉末材料:P4332(低温有序相)和Fd-3m(高温“多晶”相)。如图6所示,拉曼图谱对比清楚地展示了两种相结构的差异,而红外光谱对比区别不如拉曼明显,但依然存在细微差异。此外,可以定量地使用624和589 cm-1两个谱带的强度比来估算该材料中两相各自所占的比例。
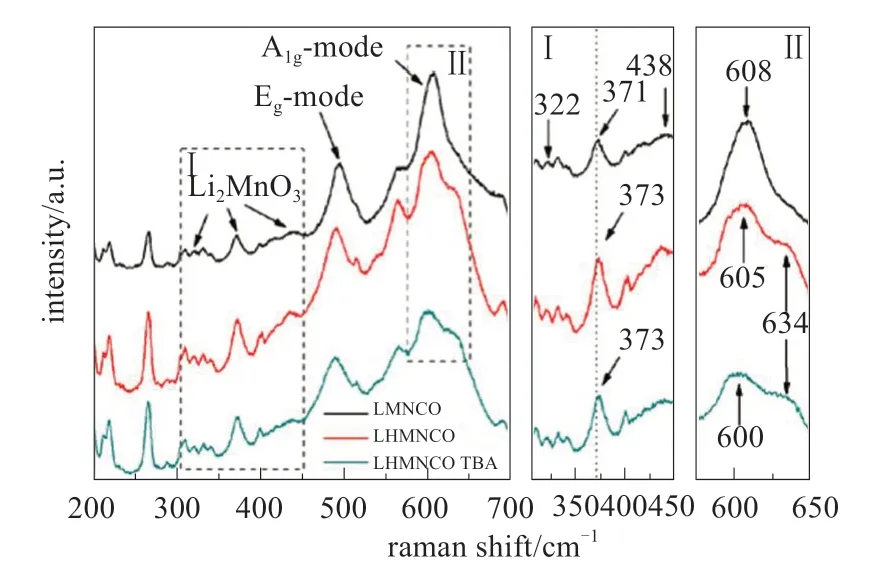
图7 原始富锂层状LMNCO和离子交换LMNCO材料的拉曼光谱[15]Fig.7 Raman spectra of ion-exchanged LHMNCO and LHMNCO TBA powders in comparison with those of pristine Li-excess layered LMNCO[15]

图8 Co3O4(I) 、S-LiCoO2(II)及 L-LiCoO2 (III)纳米片阵列的拉曼谱图[11]Fig.8 Raman spectra of the Co3O4,S-LiCoO2 and L-LiCoO2 nanosheet arrays[11]
由于在层状结构中涉及Mn-O键的振动模型与尖晶石相中的振动模型不同,拉曼光谱也用于确定尖晶石结构的形成,即定性确定相转变。ZHAO等[15]利用拉曼光谱对层状富锂材料离子交换后向尖晶石转变进行研究。如图7所示,原始的LMNCO在322、372和431 cm-1处显示峰,这是单斜晶Li2MnO3组分的典型峰。在486和608 cm-1处的两个强峰可分别归因于弯曲Eg和拉伸A1g模式。在H+-Li+交换后,LHMNCO样品中出现634 cm-1的肩峰,这可归因于立方尖晶石相中MnO6八面体中的Mn-O振动,揭示了相变的开始。Li2MnO3在372 cm-1处的特征峰略微出现红移,表明该组分在酸性处理过程中发生化学活化,其中Li2MnO3在Li+-H+离子交换过程过程中倾向于生成Li2O并变成MnO2。LIANG等[11]使用Co3O4纳米片阵列作为模板,制备了层状LiCoO2纳米片阵列,在制备过程中还得到未退火的尖晶石LiCoO2纳米片阵列,图8证实制备LiCoO2纳米片阵列过程中,由尖晶石-尖晶石层相转变。
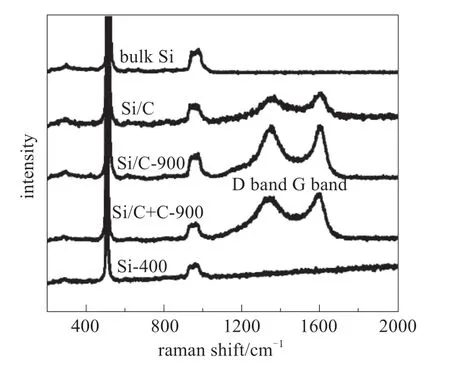
图9 不同制备条件下样品的拉曼光谱[18]Fig.9 Raman spectra of different samples[18]
对于锂离子电池负极材料来说,也可以通过光谱来研究样品表面的组分和结构,常见材料如碳基负极[16-18]、硅基负极[18-19]、钛基氧化物[20-21]、锡基氧化物[22]等。李泓等[18]制备了多孔碳硅复合负极材料,并采用拉曼光谱分别表征不同反应条件下制备的材料的组分,如图9所示,所有样品在515.2和965.5 cm-1的两个谱带都可归属于 Si 的特征振动峰。对于碳材料,代表无序相的拉曼特征峰(D峰,1350 cm-1)与代表有序相的特征峰(G峰,1590 cm-1)的比值对于结构研究很有意义,其比值的变化在一定程度上揭示了材料结构的变化。在这里,IG/ID比值与高温碳化后碳层的石墨含量成正比。
MAO等[17]使用纳米CaCO3模板热解明胶制备了富氮中孔碳材料,并通过拉曼光谱探究了充放电过程中其结构的变化(图10)。D峰与G峰的比值La值在放电期间减小,随后在充电期间增加,意味中孔碳的氮掺杂石墨烯层的微晶尺寸在Li+嵌入时增大,随后在Li+脱出时恢复。这种可逆的过程确保了材料具有良好的循环可逆性。另外D峰随着充放电过程半高宽增大,分析可能是在放电过程中,C=N键可能会部分断裂并与锂反应,最终导致碳的无序相增加。

图10 在不同电位下700 ℃热解明胶的拉曼光谱(a)和La值(b)[17]Fig.10 Raman spectra (a) and La (b) of the 700 ℃pyrolyzed gelatin at different potentials (Li+/Li)[17]
3.1.2 电解质表征
电解质是电池的重要组成部分,作用是通过电池内部在正负电极之间传输离子。它对电池的容量、工作温度范围、循环性能以及安全性能等都有重要影响。锂离子电池常见电解质为液体,一般采用锂盐的有机溶液作为其电解质。电解质锂盐不仅是电解质中锂离子的提供者,其中的阴离子也是决定电解质物理和化学性能的主要因素[23]。锂盐和溶剂间的相互作用在一定程度上也会影响电化学循环性能。王兆翔等[24]利用拉曼和红外光谱对溶剂PC和锂盐LiN(CF3SO2)2(简写为LiTFSI)之间的相互作用及溶液中离子种类进行研究。由图11(a)和11(b)可知,随着溶液中盐浓度增加,PC游离分子(710 cm-1,Raman)强度减小,并出现被指认为锂盐与PC分子相互作用的新峰(722 cm-1,Raman;1404 cm-1,IR)。而图11(c)的812 cm-1峰向高波数移动则说明阴离子和溶液中的溶剂/阳离子的相互作用增强。图11(d)则表示阴离子光谱变化是由于溶液中阳离子和阴离子(如Li+-PC-TFSI-)相互作用所导致。

图11 (a)具有盐浓度的OCH2摇摆振动的红外光谱的演化;(b)基于拟合结果的游离和相关PC分子的拉曼强度浓度依赖性;(c)具有盐浓度的SN拉伸模式的拉曼光谱演变;(d)自下而上:纯PC、不同浓度盐溶液和纯LiTFSI的SO2振动的拉曼谱[24]Fig.11 (a) evolution of the IR spectra of the OCH2 wagging mode with salt concentration; (b) concentration dependence of the Raman intensities of free and associated PC molecules based on the fitting results in Table 1; (c) evolution of the Raman spectra of the SN stretch mode with salt concentration; (d) Raman spectra of SO2 vibrations at different salt concentrations:pure PC and concentrations of 0.52,1.53,2.42 and 3.93 mol/L and pure LiTFSI,from bottom to top[24]

图12 (a)LiTFSI-H2O体系在不同 LiTFSI 浓度下的拉曼光谱和纯固体LiTFSI盐;(b)盐浓度对744 cm-1拉曼振动的影响[30]Fig.12 (a) Raman spectra of LiTFSI-H2O systems at different LiTFSI concentrations and pure solid LiTFSI salt; (b)Progression of Raman vibration at -744 cm-1 with salt concentration[30]
电解质中的溶剂参与氧化还原反应,但却不一定完全参与反应,如碳酸乙烯酯(EC)被选择性还原的最早证据来自于ZHUANG等[25]的研究;而碳酸丙烯酯(PC)分子在溶液中也因是否有Li+而有不同的优先生成物取向[26-27];不同种类溶剂[碳酸酯类与二甲基乙烷(DME)]在充、放电的某一过程中会发生不可逆反应[28]。另外,电解液在阴极表面反应后可能会产生某些荧光物质,这些具有强荧光的物质被证明是电解质分解产生的卤代磷酸锂[29]。传统电池的电解液以水为溶剂,其工作电压不超过2 V,但由于锂离子电池的工作电压高于2 V,故需要采用非水电解液体系作为其电解液。而SUO等[30]将锂盐LiTFSI直接溶于水中,制备出一种“盐水”电解质,并利用光谱法对溶液中离子相互作用进行表征。图12为LiTFSI-H2O体系不同浓度下的拉曼谱及其放大图。图中显示出最强峰(S-N-S弯曲振动)随着浓度增加向高波数转移的趋势,作者认为这是由于此种振动模式对阴离子环境变化非常敏感,并将其归因于在高浓度溶液中形成的渗滤TFSI-阴离子网。
3.1.3 SEI膜
对于电极表面的电解质被还原形成固体电解质层(SEI) 的探究,一直是研究人员关注的重点。SEI对电池的不可逆容量、循环性能、嵌锂稳定性都有重要影响。AURBACH等[31]利用傅里叶变换红外光谱表征不同电解质溶剂在电极表面生成的SEI膜的不同组分。测试中发现,溶剂为FEC其电极的吸收率高于溶剂为EC,这说明在FEC中形成了较厚的表面膜。另外从光谱中得出结论(图13),电极在FEC中其表面形成的SEI膜组分里含有大量的LiPF6分解后的产物。许康等[32]通过FTIR分析鉴定在电极表面上形成的固体产物即SEI层,如图14所示,证实SEI组分中不仅包括先前确定的甲基碳酸锂和甲醇锂,还包括草酸锂和可能的草酸甲酯。

图13 Li电极在1 mol/L (a) LiPF6/FEC/DMC和 (b)LiPF6/EC/DMC电解液中经过长循环之后表面的FTIR[31]Fig.13 FTIR spectra of Li electrode surface after prolonged cycling in (a) 1 mol/L LiPF6/FEC/DMC and (b) 1 mol/L LiPF6/EC/DMC electrolyte solutions[31]

图14 化学合成的甲基碳酸锂(CH3OCO2Li)的FTIR光谱。采用分子轨道从头算HF/6-31G (d,p)方法得到的碳酸甲基锂(CH3OCO2Li)及其二聚形式的分子结构分别显示在插图(a)和(b)中[32]Fig.14 FTIR spectrum of chemically synthesized lithium methylcarbonate (CH3OCO2Li).The molecular structures of lithium methyl carbonate (CH3OCO2Li) and its dimerized form obtained from ab initio MO calculation at HF/6-31G(d,p) level are shown in inset (a) and (b),respectively[32]

图15 在0.5 mol/L LiClO4/THF中AN和VA (10%)的SNIFTIRS光谱细节[33]Fig.15 Detailed SNIFTIRS spectra of AN and VA (10%) in 0.5 mol/L LiClO4/THF[33]
电解液中的少量成膜添加剂,能使电解液在电极表面首先发生反应,反应后的产物附着在电极表面,形成一层性质优良的SEI膜。SANTNER等[33]通过电化学红外光谱法对THF基电解液中,添加剂丙烯腈(AN)在GC表面形成SEI膜的反应进行了研究。图15是归一化的傅里叶变换红外光谱图,分别显示了在0.5 mol/L LiClO4/THF中的AN-亚乙烯基化合物(970 cm-1)的C-H变形振动的减少,及0.5 mol/L LiClO4/ THF中的AN-饱和(2242 cm-1)的增加和腈的不饱和(2225 cm-1)C≡N伸缩振动的减少。也就是说,电位降低的同时,不饱和睛类C-H键的变形振动,以及不饱和C≡N的拉伸振动减弱,而饱和C≡N的拉伸振动增强。这证明添加剂(AN)在负极表面发生还原反应。
CORTE等[34]利用原位红外技术,研究薄膜非晶硅电极界面不同组分电解质对SEI生长和厚度的研究。从图16中看出SEI膜形成的一般趋势,从界面的吸收谱中能够定性地推断SEI膜致密且均匀:由于吸收谱线存在清晰且尖锐的峰值,即电解质在电极表面均匀地移动。另外,底部的电解质谱线与原位光谱的对应一致性也说明电化学循环期间,电极表面处形成均匀致密的SEI膜。
CORTE等认为,SEI膜的厚度可以通过对光谱的定量分析获得。也就是说,可以通过分析由SEI除去溶剂后的吸收峰,以及与在相同条件下获得的电解质光谱的定量比较来获得。由此观点得到计算SEI膜厚度的等式:dSEI=kmdp/2,其中dp是电解质中消逝波的探测深度,km是锂脱嵌期间吸收峰值与电解质峰值的比。根据等式和光谱计算的值及其相应函数,得图17,进而得出结论:①锂脱嵌循环期间有着类似振幅的厚度波动,且不完全可逆;②在首周循环结束时,在1 mol/L LiClO4(PC为溶剂)中形成的SEI膜厚度值约为5 nm,而在1 mol/L LiPF6(溶剂为PC∶EC∶3DMC,添加剂为2% VC,10% FEC)中形成的SEI膜厚度值约为18 nm;③SEI膜在首周循环中大量形成,随后的循环中增长速度趋于平缓。
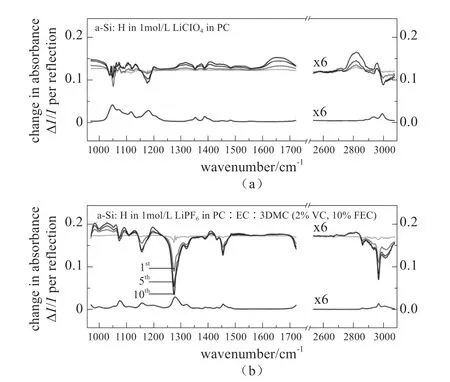
图16 薄膜非晶硅电极分别在 (a)1 mol/L LiClO4(PC为溶剂)和 (b)1 mol/L LiPF6(溶剂为PC∶EC∶3DMC,添加剂为2% VC,10% FEC)电解液中,循环1、5、10周的原位衰减全反射FTIR图谱[34]Fig.16 (a) ATR-FTIR spectra obtained in the in situ cell in 1 mol/L LiClO4 in PC after 1,5,and 10 cycles; the spectrum of the electrolyte is drawn at the bottom; (b) ATR-FTIR spectra obtained in the in situ cell in 1 mol/L LiPF6 in PC:EC:3DMC(2% VC,10% FEC) after 1,5,and 10 cycles[34]

图17 首周电化学循环期间记录的光谱估算SEI厚度和在随后循环期间硅上锂脱出完成后的SEI厚度[34]Fig.17 Thickness estimated based on spectra recorded (a)during the first electrochemical cycle and (b) at the end of silicon delithiation during the following cycles[34]
由于SEI的含量一般比较低,除以上介绍的常规方法表征外,对其组分和结构的表征常采用SERS等特殊方法进行表征,具体见3.2.2节(表面增强拉曼与红外表征)。
3.1.4 黏结剂表征
黏结剂对于以有机溶剂作为电解液的锂离子电池来说必不可少,常见的黏结剂为聚偏二氟乙烯(PVDF)。MAAZI等[35]利用FTIR证实PVDF与聚(醋酸乙烯酯)(PVAc)两种聚合物的分子间相互作用,以及这种共混物对LiMn2O4正极材料电化学性能的优异性(图18)。另外,对于LiFePO4/C电极,聚四氟乙烯(PTFE)作为黏结剂有更优的电化学性能,并且制备成本较低[36];对于Si阳极材料,聚(1-芘甲基丙烯酸甲酯)(PPy)比PVDF有着更优异的黏附性[37]。然而,PVDF作为黏结剂对某些电极材料(如LiCoO2)存在不利影响[38]。因此,出现了一些作为PVDF的替代,并对电极材料有相同作用的黏结剂,如羧甲基纤维素(CMC)[39]、结兰胶[40]等;水溶性的黏结剂,如黄原胶(XG)[41]、LA132[主要共聚物为聚丙烯腈-聚丙烯酸(PAN-PAA)][42]等;适用于Si基电极的新型黏结剂,如羧甲基壳聚糖(C-壳聚糖)[43]、羧甲基胡芦巴胶(CFG)[44]、三嵌段共聚物聚多巴胺-聚丙烯酸-聚氧乙烯(PDA-PAA-PEO)[45]等。
3.2 特殊方法表征
3.2.1 原位表征
常规的电化学研究方法以电信号为激励手段和检测手段,难以准确鉴别复杂体系的各反应物、中间物和产物并解释电化学反应机理。原位测试可以解决上述弊端,并能够得到更准确的反应产物信息[46-48],有时还可对充放电过程中Li的脱嵌行为进行表征[49-51],同时从谱学角度证明整个反应过程的可逆性[52-54]及中间产物[52-53],并间接验证非原位拉曼谱研究中所得结论。BADDOURHADJEAN等[55]利用原位拉曼技术,研究了LixV2O5薄膜在液体电解质中的首周电化学锂脱嵌,进而导致的中间相变。图19显示纯相V2O5在第一次充电过程(即锂嵌入)中发生的反应及局部结构的相变,随后在放电过程(及锂脱出)中完全恢复其初始结构,这证明其具有优异的电化学可逆性。另外在拉曼谱放大图中证明结构反应来自于反应过程中产生的固溶体,而不是通常理解的锂含量范围连续增加(0≤x≤0.15、0.3≤x≤0.7、0.9≤x≤1)而导致的连续(α、ε、δ相)相变;且与之前的研究不同,0.7≤x<1范围内未出现通常认为的δ相。

图18 PVDF、PVAc和PVDF/PVAc (1∶3)分别与LMO/C混合后电极材料的FTIR光谱[35]Fig.18 FTIR spectra of the LMO/C/PVDF,LMO/C/PVAc and LMO/C/PVDF25/PVAc75 electrodes[35]
在充电过程中Li离子发生还原反应,会伴随着部分金属锂析出,所形成的锂枝晶被认为是影响锂离子电池安全性和稳定性的关键问题。Li枝晶的生长机制与离子迁移、电极反应和SEI膜的形成有关。拉曼光谱可以用来探测离子分布和耗尽对Li沉积和形貌的影响,并用来检测局部锂浓度。由于传统拉曼光谱仪的采集信号弱且时间分辨率差,因此很难跟踪电化学反应过程中界面处的电解质浓度变化。与传统拉曼光谱仪相比,受激拉曼散射(stimulated Raman scattering,SRS)显微镜因其具有高灵敏度(< 0.5 mM)、快速成像速度(每像素约2 μs)、精细空间分辨率(300~500 nm)、无标记性质和固有3D光学切片等优点,已广泛应用于生物医学研究,但在材料和能源领域的研究报道较少。最近,CHENG等[56]利用原位SRS成像技术定量捕获Li表面离子浓度的快速演变以及浓度与Li枝晶生长的相关性(图20),通过SRS显微镜对Li离子的动态耗尽和Li枝晶的生长进行成像,分辨率为10 mM、500 nm,每帧 < 1 s。此外,这项工作对局域离子分布、Li生长速率和电压之间的相关性进行了可视化及定量化研究,揭示了Li沉积过程中存在三个动态阶段:当表面离子未耗尽时,Li呈苔藓状缓慢沉积;在部分浓度减少期间苔藓状Li和Li枝晶混合生长;当离子浓度完全耗尽后以枝晶形式快速生长。通过Li表面上的非均相Li+耗尽可以很好地解释三阶段行为。

图19 LixV2O5薄膜电极的首周充放电过程的原位拉曼光谱(a)和首周充放电过程中的原位拉曼光谱图放大图(b)[55]Fig.19 In situ Raman spectra series of the first discharge-charge process (a) and detailed view of the in situ Raman spectra collected during the first discharge; (b) of a LixV2O5 thin film electrode.0≤ x≤ 0.94.(*) LiClO4/ACN bands[55]

图20 (a)3D图像;(b)平均Li+浓度远离Li表面5 μm;(c)Li/Li对称电池在1.3 mA/cm2下的电压分布;(d) Li的增长率和标准差与时间的关系;(e) 平均[Li+]对距离电极表面5 μm,10 μm和50 μm的时间;(f)代表性的2D SRS图像;(h) 沿着 (f)(黑线)中的SRS 2D图像的x方向的1D [Li+]分布;(h) 沿着电极轮廓相对于位置(y方向)在Li表面上外推[Li+]([Li+]0 μm)[56]Fig.20 (a) 3D images showing depletion of ions near the Li surface at a current density of 4.2 mA/cm2; (b) average Li+ concentration 5 μm away from the Li surface in (a); (c)voltage profile of a Li/Li symmetric cell at 1.3 mA/cm2; (d) Li growth rate and standard deviation vs.time; (e) average [Li+]vs.time at 5 μm,10 μm and 50 μm away from the electrode surface; (f) representative 2D SRS images at 11 min,21 min and 31 min; (g) 1D [Li+]profile along the x-direction of the SRS 2D images in (f) (black lines); (h) extrapolated [Li+]on the Li surface ([Li+]0 μm) along the electrode contour versus the location (y-direction)[56]

图21 在10 MW激光功率下的原位SERS (a)LiNiMnO 和 (b)LiNiMnO[60]1.20.20.620.50.52Fig.21 In situ SERS at 10 MW laser power for (a)LiNiMnO and (b) LiNiMnO[60]1.20.20.620.50.52

图22 Li[NixLi(1-2x)/3Mn(2-x)/3]O2 充放电过程中各材料表面的反应机理(上)和充放电的不同过程(下)[60]Fig.22 Proposed surface reaction mechanism of Li[NixLi(1-2x)/3Mn(2-x)/3]O2 for oxygen activating members during charging and discharging (top) and charging/discharging curve showing the different process (bottom)[60]
3.2.2 表面增强拉曼与红外光谱表征
SERS既可表征反应完成后电极表面的产物[57],也可与原位技术同时使用表征充放电过程中产生的少量中间体[58-59]。HY等[60]利用原位表面增强拉曼光谱技术对Li[NixLi(1-2x)/3Mn(2-x)/3]O2(0≤x≤0.5)(LLNMO)一系列材料进行测试,研究在电化学循环期间电极表面物质的形成(图21)。之后与中间相碳微米球组成全电池进行原位SERS测试,以确认电解质溶液内在电极两侧的变化。结果表明,在富锂正极材料上形成Li2O,且在负极上形成LiOH∙H2O。在达到4.5 V平台时氧被活化,Li2O的形成及其随后的反应导致电解质的水解,并且在放电时溶液内变化导致Li2CO3和其他相关产物的形成(图22)。综合上述结果,HY认为,在研究中不仅应关注表面包覆以改善稳定性,同时也应关注电解质提供改善材料性能的途径[60]。

图23 (a) 吸附在Au(111)表面上的4-PBT的潜在依赖EC-TERS光谱;(b)4-PBT修饰的Au NP表面的潜在依赖性SERS光谱[61]Fig.23 (a)The potential dependent EC-TERS spectra of 4-PBT adsorbed on Au(111) surface; (b) The potential dependent SERS spectra of a 4-PBT modified Au NP surface[61]
任斌等[61]对表面增强拉曼光谱仪进行改进,自制并使用电化学尖端增强拉曼光谱(electrochemical tip-enhanced Raman spectroscopy,EC-TERS)表征固-液界面。如图23所示,可知单端键合物质在高频侧(1605 cm-1)出现主峰,在低频侧(1592 cm-1)出现肩峰;而质子化物质和双端键合物质都仅能出现单峰(1605 cm-1),上述结论在密度泛函理论计算中得到准确证实。这表明,EC-TERS比SERS更能准确反映分子结构微小变化。随着检测灵敏度的进一步提高,将有望达到电化学界面的纳米级光谱成像[61]。
由于红外光谱本身灵敏度较高,而且通常有机官能团的红外活性较强,故表面增强红外光谱一般用于痕量有机基团的测试,如生物、医学等领域的特殊研究中,在锂电池中的应用较少。
3.2.3 联用技术表征
拉曼光谱和红外光谱各有其优势,但也同时存在一定的局限性,在实际应用和分析中联用技术可以最大限度地发挥不同仪器的优势,从而使得表征结果更加全面。下面以几种联用为例,介绍其在表征方法上的特点和优势。
在3.2.2节中提到的尖端增强拉曼(TERS),实际上,此技术是扫描探针显微镜(scanning probe microscope,SPM)与表面增强拉曼光谱(SERS)的联用。同步辐射技术与拉曼/红外的联用一般用于生物、考古等特殊领域的研究,在锂电池中的应用较少。
原子力显微镜(AFM)与拉曼联用(AFMRaman)。SERGEY等[62]利用AFM-Raman联用技术研究了化学气相沉积合成锂-石墨纳米管(LGN)。由高分辨率拉曼谱可知样品具有少量的紧密凝聚的LGN层,因此推断表面有凹凸不平的多层管状结构,这一结果在同一样品区域的AFM图像中也得到验证。
热重(TG)与傅里叶变换红外光谱联用(TG-FTIR)。WANG等[63]利用TG-FTIR技术,分析了金属氧化物修饰的石墨烯复合材料并掺入环氧基质以及纯环氧树脂(EP)的热分解过程。由图24可以看出,复合材料的热分解过程类似于纯EP,特征峰出现在3600~3700 cm-1、2750~3150 cm-1、2250~2400 cm-1、1400~1650 cm-1和1100~1300 cm-1的区域中,并使得分解产物中的有机挥发物含量明显降低。

图24 (a)纯EP;(b) 石墨烯/EP;(c)SnO2-石墨烯/EP和(d) Co3O4-石墨烯/EP复合物的热解过程中气态挥发物的3D图[63]Fig.24 The 3D diagrams of the gaseous volatiles during pyrolysis process of (a) pure EP,(b) graphene/EP,(c) SnO2-graphene/EP and (d) Co3O4-graphene/EP composites[63]
气相色谱(GC)/质谱(MS)与拉曼和傅里叶变换红外光谱同时联用,由于GC-MS能够检测反应系统生成的气体和液体产物,但就液体中的痕量产物来说,Raman和FTIR光谱可以达到比GC-MS更高的灵敏度和信噪比[64]。WANG等[64]将正极材料LiCoO2包括商品的、纳米尺寸的、裸露的和Al2O3包覆的都浸泡在电池级的加有或未加LiPF6的碳酸乙烯酯(EC)和碳酸二甲酯(DMC)的混合物中,利用GC-MS、拉曼以及红外光谱表征手段,研究了由浸泡所引发的气体、液体和固体产物的组成。由图25可知,由于溶剂的分解,产生了一系列的气体产物(CH4,CH3-CH3,CH2=CH2,CO,CO2,O2和H2O等),同时在电极表面伴随着表面膜沉淀生成,其中含有Li2CO3,其中的Li+来自于LiCoO2,从而导致其结构退化。

图25 浸泡于EC/DMC溶剂的LiCoO2所产生气体种类的典型的色谱 (a)和质谱 (b)图;溶剂浸泡LiCoO2的新鲜的 (c)和陈化的(d)固体的红外光谱;溶剂浸泡LiCoO2在以DMC清洗前(e)和清洗后(f)并在手套箱中保存7后其表面沉积物的Raman光谱[64]Fig.25 Typical (a) GC and (b) MS spectra of gas species generated on LiCoO2 soaked in EC/DMC; IR spectra of the (c) fresh and (d) aged solids from solvent-soaked LiCoO2; Raman spectra of the deposits on solvent-soaked LiCoO2 (e) before and (f)after rinsing with DMC and then stored in the glove box for 7 days[64]
扫描电子显微镜(SEM)与拉曼联用(SEM-Raman)。在3.2.2节中,HY等[60]利用原位表面增强拉曼研究了电化学循环期间电极表面物质的形成。与此同时,HY还利用了SEM-Raman方法,证实表面物质Li2CO3在表面的分布。图26中,看到Li2CO3在两种电极表面均存在,但覆盖区域不同,可以从拉曼映射(Raman Mapping)图像中清楚看出,相比于LiNi0.5Mn0.5O2表面,Li1.2Ni0.2Mn0.6O2表面表现出了更高的覆盖率(黑色区域为背景信号)。
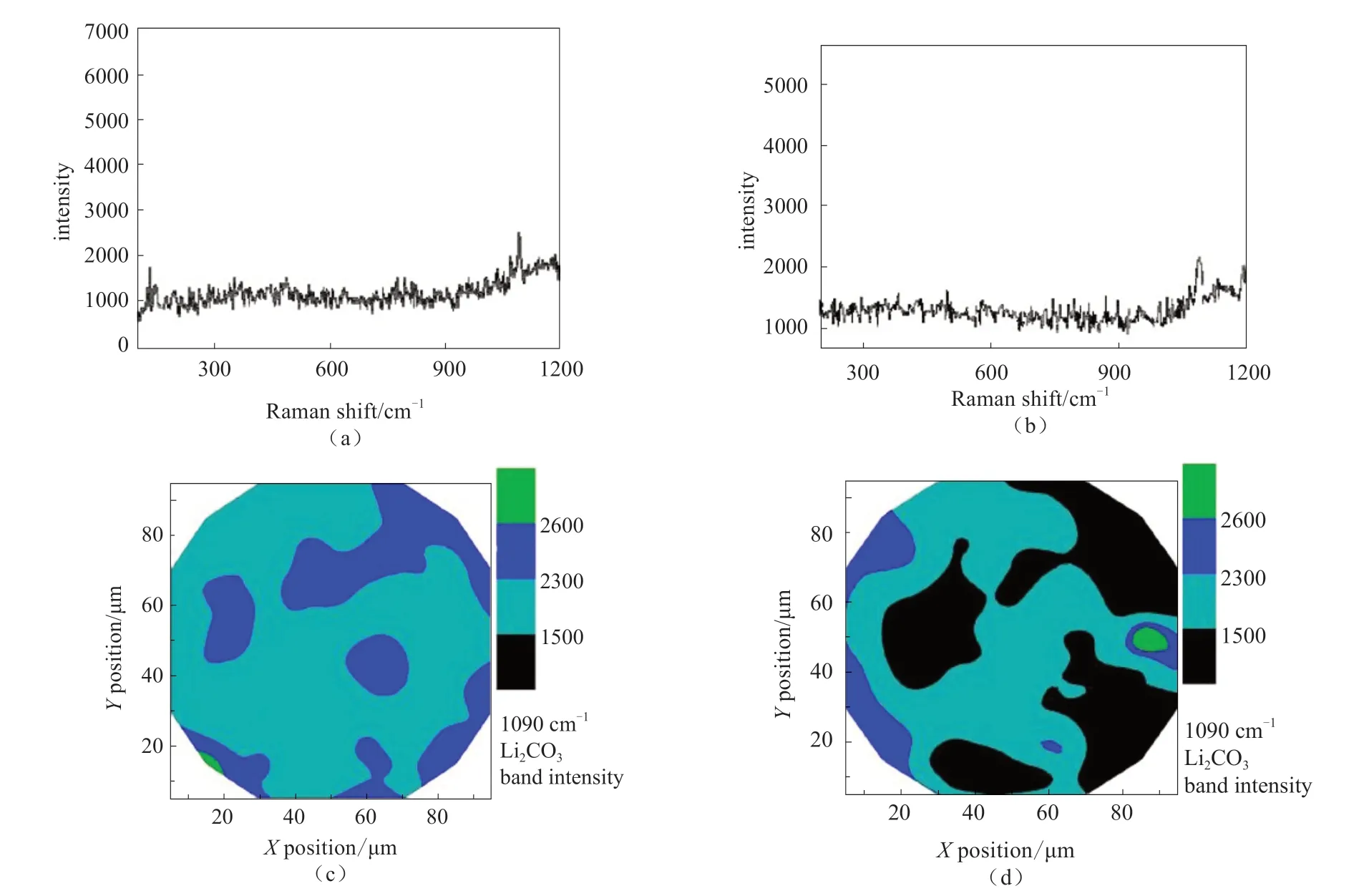
图26 (a)Li1.2Ni0.2Mn0.6O2和 (b) LiNi0.5Mn0.5O2在第1次循环放电至2.5 V后的拉曼光谱;(c) Li1.2Ni0.2Mn0.6O2和 (d)LiNi0.5Mn0.5O2在放电至2.0 V后的拉曼光谱显示出1090 cm-1的Li2CO3振动强度[60]Fig.26 Example of Raman spectra of (a) Li1.2Ni0.2Mn0.6O2 and (b) LiNi0.5Mn0.5O2 during mapping after the 1st cycle discharged to 2.5 V;Raman mapping of (c) Li1.2Ni0.2Mn0.6O2 and (d) LiNi0.5Mn0.5O2 after discharge to 2.0 V showing the 1090 cm-1 Li2CO3 band intensity[60]

图27 使用常规固体电解质的(a)带电和(b)放电的LiCoO2电极层的光学显微照片;(a)和(b)的显微照片的拉曼映射图分别显示为(c)和(d)[65]Fig.27 Optical micrographs of the (a) charged and (b)discharged LiCoO2 electrode layers using conventional solid electrolyte; Raman mapping images for the micrographs of(a) and (b) are shown as (c) and (d),respectively[65]
在全固态电池中,利用Raman Mapping还可以反映界面处反应物及反应位点。OTOYAMA等[65]使用常规或较小的固体电解质在块状全固态锂电池中对充电和放电的LiCoO2复合正电极层进行拉曼映射。如图27所示,映射图像表明,在使用较小的固体电解质颗粒的情况下,由于LiCoO2和固体电解质之间的充分接触,发生了均匀的充电/放电反应。从拉曼成像中可以看出,使用较小的固体电解质颗粒是获得全固态锂电池中复合正电极均匀反应分布的有效方法。
衰减全反射(attenuated total refraction,ATR)与红外连用(ATR-FTIR)。通过样品表面的反射信号获得样品表层有机成分的结构信息,且可消除样品中水分对测试结果的影响。SAQIB等[66]使用ATR-FTIR定量原位测量锂-硫电池电极之间的多硫化锂含量。如图28所示,多硫化物的有序和浓度与特征红外吸收带的位置和面积呈正相关。

图28 (a)DMSO和(b)TEGDME中多硫化锂的ATR-FTIR光谱[66]Fig.28 ATR-FTIR spectra of lithium polysulfides in (a) DMSO,and (b) TEGDME[66]

表5 通过拉曼/红外光谱可获得的信息Table 5 Result obtained by Raman/infrared spectroscopy

表6 一些锂离子电池电极材料的主要拉曼峰及其振动模式Table 6 Wavenumbers of Raman-active modes of some important cathode and anode materials of the lithium-ion battery

表7 部分锂离子电池电解质官能团的主要红外峰及其振动模式[78-79]Table 7 Wavenumbers of FTIR-active modes of some important electrolytes functional group of the lithium-ion battery[78-79]
4 结 语
拉曼光谱和傅里叶变换红外光谱是重要的物理表征方法,在电化学领域尤其是锂电池领域具有广泛的应用,如分子价键、官能团振动和转动能级跃迁状态、物相结构变化、稳定性、表面现象以及反应机理,相关谱学数据又可以计算化学键的键能、键长、键角等,通过光谱可获得的信息具体见表5。正确且合理使用两种表征方法可以帮助研究人员更深入理解电池,从而提升研究成果的权威性。表6列出部分电极材料的主要拉曼峰及其振动模式,表7列出部分电解质官能团的主要红外峰及其振动模式。
然而,尽管拉曼光谱和红外光谱在锂电池物理表征中具有特殊重要的地位,但准确全面地分析实验所获光谱包括相关振动峰指认,还需要大量的文献积累和对所测样品及测试仪器的深入了解。以拉曼光谱为例,拉曼光谱实验与分析中存在不易确定的因素主要有以下九类:①样品本身的均匀性会影响测试结果的可重复性;②由于激光聚焦深度的微弱差异使得信号采集镜头对应的光谱散射截面不同;③由于可能的局域功率密度过高导致的温升对一些热稳定性比较差的材料的热稳定性造成影响;④由于阻隔滤波片质量下降影响低波数信号的采集;⑤由于测试谱仪的分辨率限制或激光器本身出现杂散光等仪器设备性能下降对真实结果造成干扰;⑥测试中样品的荧光效应会对拉曼光谱的采集造成不同程度的背景干扰,此时弱峰可能会湮没在大的荧光背景中;⑦由于空间中随机出现的宇宙射线对信号采集的干扰;⑧分峰拟合过程中存在的不确定因素(特别是涉及到多峰的分峰拟合谱仪的光谱分辨率较低时)会增加拟合结果的不确定性;⑨由于缺乏全面系统的谱图数据库,使得一些测试结果中的振动峰难以指认,而红外光谱具备全面完整的谱图数据库,故不存在这方面问题。不仅如此,有时对于材料晶体结构的研究或者物质官能团的鉴定,仅有此两种表征及其峰位指认是不够的,还需要辅助一些其它表征手段,如X射线衍射(XRD)、核磁共振(NMR)、深紫外光谱(UV)等的配合来进行综合分析,并合理地运用一些联用技术完善实验结论或结合理论计算和模拟进一步加以验证。
猜你喜欢
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
陶瓷学报(2021年1期)2021-04-13 01:33:40
电子测试(2018年18期)2018-11-14 02:30:36
中学生数理化(高中版.高二数学)(2017年1期)2017-04-16 05:33:49
光学精密工程(2016年1期)2016-11-07 09:01:00
电源技术(2015年9期)2015-06-05 09:36:06
应用化工(2014年11期)2014-08-16 15:59:13
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:38
化学分析计量(2013年1期)2013-03-11 16:37:15
物理与工程(2013年1期)2013-03-11 16:03:39
