
28家药品生产企业飞行检查和跟踪检查问题分析及监管对策
2019-09-10 07:22陆仕华韦莹莹李杉韦广辉
中国药房 2019年20期
陆仕华 韦莹莹 李杉 韦广辉
中圖分类号 R95 文献标志码 A 文章编号 1001-0408(2019)20-2741-05
DOI 10.6039/j.issn.1001-0408.2019.20.02
摘 要 目的:为药品生产企业完善《药品生产管理规范》(GMP)质量管理体系建设和药品监管部门提高监管水平提供参考。方法:通过对2018年2月6日-2019年1月25日国家药品监督管理局网站公布的药品飞行检查和药品跟踪检查通报的28家药品生产企业存在的问题进行分析总结,找出共性问题并对其进行原因分析,进而提出监管对策。结果与结论:药品生产企业自身存在一些GMP实施不到位的问题,例如部分关键岗位人员未能正确履职、相关人员培训效果不理想,实际生产工艺与批准的法定工艺不一致、物料管理不规范、批生产记录不完整、不能对药品生产全周期进行有效监控等问题;而监管部门也存在检查员现场检查的尺度差异较大、检查员的检查能力和水平有待加强、监管手段创新不足等问题。建议药品生产企业应完善企业GMP质量管理体系建设、加强企业相关人员的培训;建议监管部门继续推进企业的“放管服”改革工作,严格检查员准入条件,加强对检查员队伍的业务培训和思想建设,加强监管制度建设,重视监管手段创新,从而共同维护药品的安全有效和质量可控。
关键词 药品监管;药品生产管理规范;生产企业;对策
ABSTRACT OBJECTIVE: To provide reference for pharmaceutical manufacturers improving the quality system of GMP and drug regulatory departments improving their supervision level. METHODS: Through analyzing and summarizing the problems existing in the 28 pharmaceutical enterprises which had been published on the website in the National Medical Products Administration from February 6th, 2018 to January 25th, 2019, the common problems were found and their causes were analyzed, then the regulatory countermeasures were put forward. RESULTS & CONCLUSIONS: Pharmaceutical enterprises have some problems of inadequate implementation of GMP, such as the inadequate performance of personnel in key positions and the unsatisfactory training effect of relevant personnel, the inconsistency between actual production technology and approved legal technology, the non-standard management of enterprise materials, the incomplete batch production records and the inability to effectively monitor the production cycle. However, there are also some problems in the supervision department, such as the large difference in the scale of inspectors’ on-site inspection, the need to strengthen the inspectors’ inspection ability and level, and the lack of innovation in the means of supervision. It is suggested that pharmaceutical manufacturers should improve the construction of GMP quality management system and strengthen the training of relevant personnel; the regulatory authorities should continue to promote the reform of “release, control and service”, strictly enforce the access conditions of inspectors, strengthen the training of inspectors and ideological construction of the inspector team,further strengthen the construction of supervision system and enhance the innovation of supervision means, so as to jointly maintain the safety, effectiveness and quality controllability of medicines.
KEYWORDS Drug supervision; GMP; Manufacturer; Countermeasures
近年来,随着国家药品审评审批制度改革以及药品监管改革的不断深入,药品飞行检查、跟踪检查已成为新时期药品安全监管的新常态,及时发现和总结新形势下药品安全监管过程中发现的缺陷问题就显得尤为重要。GMP飞行检查是对药品生产企业跟踪检查的一种形式,其重点检查对象是涉嫌违反《药品生产管理规范(GMP)》或有不良行为记录的药品生产企业[1]。本研究通过回顾和分析国家药品监督管理局飞行检查和跟踪检查过程中发现的药品生产企业存在的问题,对这些问题进行合并归纳,找出共性问题并对其进行原因分析,并针对上述问题提出监管对策,旨在为进一步切实提升国家药品安全监管能力、帮助药品生产企业在实施GMP时避免出现类似的缺陷情况提供参考。
1 资料与方法
数据资料来源于2018年2月6日-2019年1月25日国家药品监督管理局网站公布的药品飞行检查和药品跟踪检查通报,收集被通报的28家药品生产企业检查中存在的问题,并结合国家药品监督管理局数据管理系统中相关企业的信息数据进行分析。
2 结果
2.1 检查结果总体情况
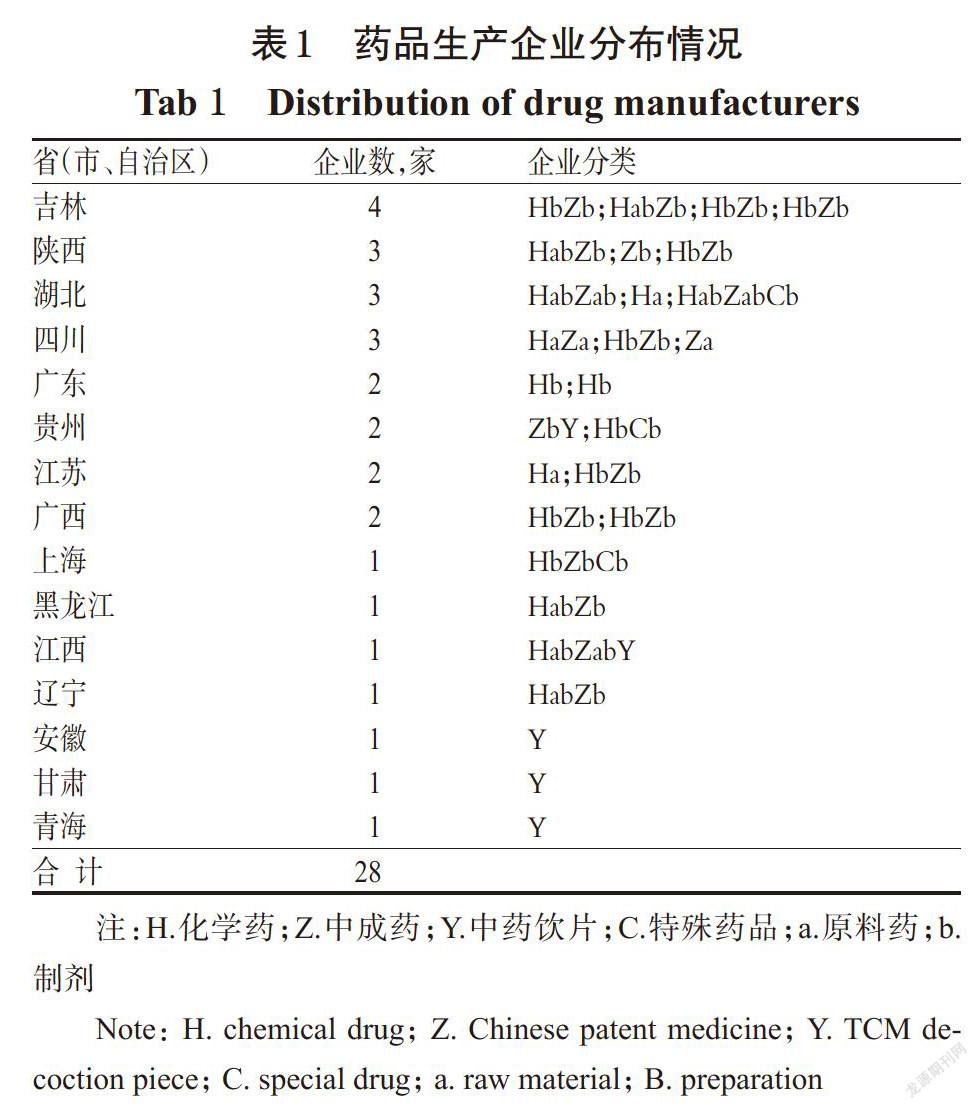
国家药品监督管理局网站公布的药品飞行检查和药品跟踪检查通报的28家药品生产企业分布于全国15个省(市、自治区)(见表1);28家药品生产企业的生产范围包括粉针剂、冻干粉针剂、小容量注射剂、大容量注射剂、片剂、硬胶囊剂、软胶囊剂、糖浆剂、颗粒剂、干混悬剂、露剂、丸剂、合剂、散剂、酒剂、酊剂、口服溶液剂、口服液、煎膏剂、膏药、流浸膏剂、口服乳剂、软膏剂、乳膏剂、贴膏剂、茶剂、锭剂、搽剂、涂剂、眼用乳膏剂、滴眼剂、滴鼻剂、滴耳剂、洗剂、冲洗剂、中药饮片、中药前处理提取物、原料药等类型。
2.2 企业检查过程中发现的主要问题分析
2.2.1 部分关键岗位人员未能正确履职,相关人员培训效果不理想 通过对28家药品生产企业存在的问题进行分析发现,部分药品生产企业关键人员培训效果不理想,部分操作没有严格按操作规程进行生产操作,还有部分企业中药材接收管理人员缺乏中药材养护知识。个别企业生产管理负责人未能严格履行职责,例如未能确保厂房和设备的维护保养,以保持其良好的运行状态;未能确保完成各种必要的验证工作;未能确保生产相关人员经过必要的上岗前培训;中间产品含量实际检测数据未达到内控标准,未进行超标结果(OOS)调查即放行充填等。
2.2.2 实际生产工艺与批准的法定工艺不一致 少部分企业存在随意变更关键生产工艺参数、不按批准的法定工艺组织生产的情况,如使用外购提取物代替企业的药材提取物进行投料,个别企业中药提取物的浸润时间与工艺规程规定的时间不一致。这些随意变更关键参数和不按法定工艺组织生产的行为严重违反了药品GMP的相关规定。
2.2.3 物料管理不规范 个别企业冷库存放的某品种浸膏货位卡中的入库数量与批生产记录显示数量不一致;个别企业部分物料超过有效期未及时处理;需阴凉储存的物料品种放在常温库;防止物料污染和交叉污染的措施不足,无法确保物料的正确存储和发运[例如洁净区原辅料暂存间供制粒用乙醇和一般区制粒用乙醇已被污染,塑料桶(外标识为“提取”)内有黑色异物,或者塑料桶密封不严];某些不合格的药材没有隔离存放;玉米淀粉、乙醇等已入洁净區暂存间台账的物料,尚堆放在一般区电梯口,未对其进行控制管理。
2.2.4 批生产记录不完整,不能对药品生产全周期进行有效监控 部分企业存在批生产记录内容不全、关键操作未在生产记录中体现的情况,如某企业某品种批生产记录中缺少领料单、称量过程记录、物料平衡检查和尾料(粗粉、细粉)记录等;某品种烘干岗位标准操作规程中规定的该品种湿品烘干前的处理步骤未在干燥工序的批生产记录中写明,操作工人实际处理过程未记录。还有部分企业存在批生产记录不真实的情况,如生产记录显示同一人同一时间段在不同车间进行操作;实际操作过程及操作时间与记录不相符等。
2.2.5 质量控制实验室管理不规范 部分企业对药材和中药饮片未进行全检,部分药材未进行含量测定、重金属及有害元素等检验即投入使用;部分中药饮片未按法定质量标准进行“检查项目”和“含量测定”检验。部分企业存在委托检验项目不全的情况,如检验报告项目不全,缺少二氧化硫、浸出物等项;个别企业存在引用供应商的检测结果代替成品检验报告的情况。部分企业试剂、对照品无购进和使用记录,如无对照品溶液配制记录(仅记录取样量)以及领用记录,同时在检验原始记录中仅记录中国食品药品检定研究院对照品的批号,未体现对照品溶液的相关信息,难以溯源。
2.2.6 计算机化系统和数据管理不规范,数据完整性欠佳 部分企业对计算机化系统的管理不符合药品GMP附录中计算机化系统要求,如紫外分光光度计、高效液相色谱仪、气相色谱仪、原子吸收分光光度计等均为单机版,均未配备审计追踪功能;高效液相色谱仪色谱工作站是N2000,版本号是4.0,无账户登录和分级权限控制,计算机系统未禁止剪切、删除、重命名等功能,系统时间亦未锁定;计算机系统无质量保证(QA)人员登录账号。部分企业数据管理不规范,如实验室未按照企业制定的《实验室原始数据的管理规定》对薄层色谱照片进行电子数据管理,仅由实验员手机拍照后打印,原始电子照片未进行统一存储和备份。个别企业未能提供检验原始记录,关键数据记录存在信息不一致、未保存、无法溯源等情况,如企业不保存物料货位卡,在物料发放结束后进行了销毁,仅使用未受控的电子文档记录;未保存生产车间设备使用日志等。
2.2.7 产品工艺验证内容不全 部分企业存在产品工艺验证内容不全的情况,如某企业生产的某品种混合粉实际生产批量与工艺验证批量不一致;某个品种工艺规程的变更无相关研究数据或验证数据支持;工艺验证每个剂型只验证一个品种,没有按每个品种批准的工艺进行验证;工艺再验证产品未进行质量评估和稳定性考察;个别企业生产的板蓝根片未对板蓝根片生产工艺进行定期再验证,未开展板蓝根稠膏储存时间验证等。
2.2.8 未取得 “双证”就进行生产销售 个别企业存在涉嫌违法销售未取得《药品生产许可证》生产的原料药,如某企业生产的盐酸小檗碱在未取得药品GMP证书之前即开始生产销售,当地药品监督管理局已对上述行为进行立案调查。
2.2.9 GMP管理与实际应用脱节 部分企业的GMP管理水平没有跟上新时期药品安全监管的新形势、新要求,存在GMP管理要求与实际应用脱节的情况。药品GMP强调的是建立覆盖整个药品生命周期的质量管理体系,要求企业人员全员参与、全过程参与[1],但在实际中企业存在操作规程和管理制度与操作人员或者QA人员实际工作不一致的情况,如个别企业的GMP办公室人员、QA人员在进行体系文件的制定时没有及时跟生产和检验一线的人员进行有效的沟通交流,存在“闭门造车”的现象,导致制定出来的体系文件、制度文件和操作规程不完全符合实际操作要求。
2.3 监管部门检查执行中存在的问题分析
2.3.1 检查员现场检查的尺度差异较大 药品GMP检查员一旦取得检查员资格,只要通过每年的年度审核就可以一直具有检查员的资格。较低的门槛会导致检查员执法水平和专业素养的较大差异,也为整个检查员队伍建设带来了困难,不能保证监管职能的有效发挥。在培训方面对检查员多采用会议培训,缺乏有针对性的培训,而这样的方式往往导致培训深度不够,缺乏技术性指导,为检查质量带来隐患[2]。其次,对GMP检查员的培训缺乏有效的动态管理机制,部分检查员因为没有优胜劣汰的压力,不重视自身能力的提升,最后也会导致检查标准尺度无法有效统一,检查过程也会存在一定的差异化[3]。
2.3.2 检查员的检查能力和水平有待加强 现有药品GMP檢查员都是由药品监督管理局系统内的人员组成,主要分布在省(市、自治区)局、市局、县局及各层级检验所[4]。另外,检查员分布于不同的岗位,在平时的工作中不一定是从事药品生产监管岗位,或者是原来在生产岗位的已经轮岗到了其他部门和岗位;有的检查员药品生产相关经验不足,其检查能力和水平参差不齐[5]。从对检查员现场检查报告和现场检查原始记录技术审核的情况看,也印证了这个问题,例如检查的原始记录不能支撑现场检查报告,甚至有的原始记录只是把现场的设备、物料和所看到的文件名称记录一下,未发现任何问题;现场检查方案中要求重点检查的内容未深入检查,工作浮于表面,未深究问题发生的根源,对缺陷项目未基于系统和风险进行分级,造成缺陷项目分级不准确,检查的质量还有待进一步提高。
2.3.3 检查员的责任感和责任心有待加强 药品监管机构对药品GMP检查员的管理和荣誉激励重视度不够,没有向检查员灌输一种使命意识,也没有具体的荣誉激励和鼓励机制,使GMP检查员没有认识到该项工作的重要性、缺乏使命感。GMP检查员的考核没有一系列完整科学的体系,以往对其考核多为培训后考核及年审[6-7],这样集中化的考核形式很难反映出检查员的实际能力,也不能有效激励检查员的学习和进步;考核的内容也过于单一,集中于技术和方法等,较少涉及能力测评和职业操守等方面内容,以致部分检查员容易出现责任心和进取心不强,甚至有应付心理或是依赖心理的情况。
2.3.4 检查员的检查时间不够充沛 目前大部分的GMP检查员一般都是在检查的2~3天前才能决定是否能参加GMP的相关检查,这也是由大部分检查员的兼职身份所决定的。GMP检查员会提前2~3天知道自己将要检查的企业,然后领取现场检查方案,了解企业的基本情况和检查计划,然后通过2天多的现场检查和查阅企业资料,最后在第3天检查结束前必须完成现场检查情况的总结和现场检查报告的撰写。这样的检查方式对检查员来说时间非常紧迫,任务也非常繁重,并要在有限的时间内发现问题,对检查员的素质要求很高。
2.3.5 监管信息缺乏整合 监管部门在制订监督检查、跟踪检查、飞行检查方案时与药品抽检评价、药品稽查、药品不良反应监测和药品投诉举报平台对接不紧密,没有及时掌握相关企业相关产品的质量公告、未结案件、不良反应数据以及投诉举报等情况,没能在检查前就及时了解企业的情况,并把相关的信息制定成现场检查方案提交检查组,导致检查效率较低,不能有效地帮助企业发现问题和及时改进提升。
3 对策与措施
3.1 企业角度
3.1.1 完善企业GMP质量管理体系建设 进一步完善企业的GMP质量管理体系建设,特别是文件管理体系的制定要符合企业的实际情况,不能照搬照抄、生搬硬套GMP条款,记录和表格的设计制订也要按实际的生产工艺规程和检验规程等进行,避免存在GMP文件管理要求与实际应用脱节的情况出现,最大程度地做到既方便记录又能符合GMP的要求,最终保证生产出符合注册批准要求的药品,确保其安全、有效和质量可控。
3.1.2 继续加强对企业人员的培训 企业应加强对其人员特别是关键岗位人员的培训和考核。GMP培训过程中要充分注重培训的时效性、科学性和培训的实际效果,使接受培训的人员能真正掌握本企业的GMP质量管理体系文件和相关管理操作规程,加强人员GMP管理意识,在实际的工作中按照GMP的要求进行生产管理,做到质量管理体系实践和标准的统一,尽量规范化操作,避免在生产管理过程中出现不符合GMP要求的情况。
3.1.3 继续推进企业的“放管服”改革工作 2016年,国家药品监督管理局已把无菌药品GMP认证工作下“放”到各省局。为确保国家药品监督管理局下放无菌药品GMP认证在短时间内“接得住、接得稳”,在监管过程中就要严把GMP认证标准不放松,在接好 “放”的同时还要坚持“管”好,继续对药品生产企业进行跟踪检查和飞行检查,充分发挥飞行检查“以问题为导向、围绕风险开展检查”的检查作用,坚持“检查频次不降低、检查力度不放松”的原则,防止“证后回潮”现象[8-9]。在加强监管的同时,药品监管部门更要“服”务好企业。在本次调查中发现,被收回药品GMP证书的企业绝大多数是中小型企业,企业规模小、人才缺乏、规范程度较低,这就要求药品监管部门充分发挥技术力量,采取多举措对企业进行帮扶,提高辖区内药品生产企业的生产管理水平。
3.2 药品监管部门角度
3.2.1 严格GMP检查员准入条件 在遴选GMP检查员时,要对检查员的遴选条件进行严格限制,并对其专业技能、职业操守等方面作出具体要求。检查员的专业至少应该是医药学和相关专业背景[10],确保遴选入库的检查员是从事药品生产监管、稽查、药品检验或有过药品生产工作经历的人员,通过培训就能胜任药品GMP检查工作;同时,提高入选检查员的执法水平和专业素养,保证药品检查的质量[11]。
3.2.2 加强对检查员队伍的业务培训和思想建设 继续推行检查员继续教育制度。检查员的继续教育过程既要注重理论学习,更要注重與实践相结合。培训方式宜采取多样化模式,例如通过试点推行与企业合作共建实训基地等形式对检查员进行实训,在实训的过程中可对遇到的热点和难点问题进行分组讨论交流,对现场检查中发现的问题进行探讨,统一检查标准,制订系统的检查方法,提升检查员的检查能力和检查水平,以使其更好地适应检查工作,提高GMP认证检查质量。此外,根据《国务院关于改革药品医疗器械审评审批制度的意见》[12]要求,药品监管部门应着手推进职业化的药品医疗器械检查员队伍建设,逐步实现检查员的专职化、专业化。
药品监督管理部门还应高度重视药品GMP检查员队伍的思想建设工作,要把GMP检查工作宣贯上升为一种职责、一种荣誉,让检查员感觉到在进行GMP认证和检查时是在履行国家赋予的重要职责,增强其使命感,以此鼓舞检查员加强自身修养专业知识的学习,逐步提高操作水平,积极努力做好工作,从而增强药品的监督和检查的效果。
3.2.3 加强监管制度建设,重视监管手段创新 在监管制度上,应加强制度建设,同时以制度建设为抓手,进一步强化注册检查、稽查检查、抽样评价和不良反应监测检查的联动检查机制,形成合力;充分运用大数据平台为监管进行数据分析,确保检查方案的合理性和针对性。在监管手段上,应重视监管手段的创新,以品种为主线进行按品种的精准检查,对具体的某个药品品种进行注册、投料、生产过程的全生命周期检查,确保药品质量;同时,对有潜在风险的品种及其生产企业应及时进行质量风险评估和风险防控检查。此外,在监管过程中还可创新性地应用药品生产智慧监管模式,搭建药品生产智慧监管平台,对药品生产企业的基本情况,信息、物料的进厂接收、入库、流转、销售,物料、中间产品和成品检验等数据录入智慧监管平台进行信息化管理,分析药品生产质量数据,自动预警、分析研判风险信息,为监管部门的日常监督检查、跟踪检查和飞行检查提供精准信息[13]。通过这些创新监管手段逐步破解当前监管过程中存在的难题,做好新时期、新形势下的药品安全监管工作,增强解决问题和发现问题的能力。
综上所述,本研究通过对国家药品监督管理局网站公布的药品飞行检查和药品跟踪检查的28家药品生产企业存在的问题进行分析总结,笔者建议药品生产企业应完善企业GMP质量管理体系建设、加强企业相关人员的培训;建议监管部门继续推进“放管服”改革工作,严格检查员准入条件,加强对检查员队伍的业务培训,加强监管制度建设,重视监管手段创新,从而共同维护药品的安全有效和质量可控。
参考文献
[ 1 ] 韩达斌,刘学良,潘平,等.青海省藏药生产企业新修订药品GMP认证检查缺陷项目分析与对策研究[J].中国药事,2017,31(1):32-36.
[ 2 ] 邓婷,施健,杨成钢.我国 GMP 认证专职检查员队伍建设模式及对策研究[J].中国药事,2016,30(8):756-761.
[ 3 ] 唐文燕,张华,等.国内外药品GMP检查员培训标准体系对比[J].上海医药,2017,38(15):55-57.
[ 4 ] 屈浩鹏.胜任素质模型在药品GMP检查员培训中的应用研究[J].首都食品与医药,2015(10):7-8.
[ 5 ] 董作军,钟元华,等.我国药品GMP监管体系存在问题的研究及思考[J].中国现代应用药学,2017,34(7):1049-1052.
[ 6 ] 邹任贤,俞佳宁,颛孙燕.国内药品GMP检查员培训标准体系的探索[J].上海医药,2018,39(9):57-60.
[ 7 ] 李士高,李龙,罗京京,等.安徽省药品生产质量管理规范检查员结构现状分析[J].安徽医药,2016,20(12):2350-2352.
[ 8 ] 温松,郑凯,肖棣文.“放管服”背景下广东特殊药品行政许可制度改革:探索、挑战与思考[J].广东行政学院学报,2018,30(3):34-43.
[ 9 ] 陆仕华,韦莹莹,李杉,等.药品生产企业 GMP(2010年修订)飞行检查情况分析[J].中国现代应用药学,2019,36(2):228-231.
[10] 韩冬,赵迎欢,田丽娟.基于利益相关者视觉的医药企业药品安全责任研究[J].中国医药工业杂志,2019,50(1):129-133.
[11] 沈黎新,钟元华,沈莉,等.新版药品 GMP 实施效果调查与分析[J].中国现代应用药学,2018,35(3):436-439.
[12] 中共中央办公厅,国务院办公厅.关于深化审评审批制度改革鼓励药品医疗器械创新的意见[Z].2017.
[13] 楼双凤.探索药品生产智慧监管,筑牢药品质量安全防线[J].上海医药,2018,39(9):51-53.
(收稿日期:2019-03-11 修回日期:2019-09-24)
(编辑:孙 冰)
猜你喜欢
电脑知识与技术(2022年11期)2022-05-31
教育家(2022年18期)2022-05-13
华东理工大学学报(社会科学版)(2022年1期)2022-04-14
少儿科学周刊·少年版(2018年12期)2018-01-26
商情(2012年48期)2017-11-08
现代商贸工业(2016年14期)2016-12-27
商(2016年34期)2016-11-24
现代经济信息(2016年2期)2016-10-27
商(2016年21期)2016-07-06
中学化学(2015年5期)2015-07-13
